Introduction
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in infants and children, with approximately 350 new cases in the United States each year. It is the third most common solid tumor in children after neuroblastoma and Wilms tumor, making up 10-15% of all solid pediatric tumors.1 By definition, RMS is a sarcoma with skeletal muscle differentiation but which can occur in sites where skeletal muscle is not found, suggesting that it is a tumor of primitive mesenchymal skeletal muscle differentiation.2 RMS incidence follows a bimodal age pattern. The first window of increased incidence, which accounts for more than 50% of cases, occurs in the first decade. This period peaks at a median of 2 years of age, and consists mainly of embryonal (E-RMS) or botryoid RMS (B-RMS). The second peak occurs during adolescence and more commonly consists of alveolar RMS (A-RMS).1 In addition to varying in their age at presentation, the histopathologic variants of RMS (embryonal, botryoid, and alveolar) are also associated with different patterns of primary presentation and subsequent outcomes. E-RMS tends to occur in the head/neck and genitourinary (GU) system and the majority of patients survive beyond 5 years. A-RMS is often seen in the extremities and tends to metastasize early with a worse survival than E-RMS.3
Since its discovery in 1854, study of the clinical and pathologic features of RMS have progressively led to the development of uniform diagnostic criteria and staging systems.4 The prognostic outlook for RMS has improved significantly with time, from an estimated survival of 25% in 1970 to over 70% now.1 This success is due to effective multimodal, risk-adapted therapy, and better supportive care. Emerging information on tumor biology has afforded new insights into the pathogenesis of this tumor and has allowed advances in risk-based management.
Genitourinary RMS
Fifteen to 20 percent of all RMS cases arise from the GU system.5 The most common GU sites are the prostate, bladder, and paratestisticular. Vaginal, cervical and uterine RMS are relatively unusual sites. As with RMS in general, survival rates vary by site within the sub-group of GU-RMS. Specifically, vaginal and paratesticular RMS enjoy a better prognosis than bladder/prostate RMS.
Epidemiology/Risk Factors
Nearly 75% of GU-RMS cases are diagnosed before the age of 5 years, with a 3.3:1 male predominance.6 The age of diagnosis impacts prognosis independently from tumor histology, and patients who are <1 year and >10 years old have a worse event-free survival rate compared to those between 1 and 9 years of age (53% and 51%, vs. 71%, respectively).7 There are established risk factors for developing RMS and given the young age of those affected; these are mostly congenital as opposed to acquired risk factors. Costello Syndrome, Gorlin’s basal cell nevus syndrome, Rubinstein–Taybi syndrome, trisomy 21 syndrome, Beckwith–Wiedemann syndrome, and fetal alcohol syndrome have all been associated with RMS.8 Two genetic conditions require particular mention: (1) Neurofibromatosis is the most common syndrome in which RMS has been described. From the patients enrolled in the Intergroup Rhabdomyosarcoma Study IV (IRS IV), the incidence of neurofibromatosis was 0.5%.9 (2) The Li-Fraumeni syndrome is a germline mutation of the TP53 tumor suppressor gene and is transmitted in an autosomal dominant manner. An increased incidence of RMS has been found in association with Li-Fraumeni Syndrome as have other solid tumors in childhood, including adrenocortical carcinoma.10 Environmental factors associated with RMS include radiation exposure and prior chemotherapy with alklyating agents. The increased risk of secondary malignancies in those patients with neurofibromatosis and Li–Fraumeni syndromes after treatment with alkylating agents and radiation therapy for RMS draws a link between genetics and environment. It is therefore desirable to limit the use of alkylating agents and radiotherapy in these particular populations.11
Tumor Biology/Genetics
The histology of RMS consists of small round blue cells, with varying numbers of spindle cells resembling fetal skeletal muscle. The Intergroup Rhabdomyosarcoma Study Group (IRSG) has developed a pathology classification recognizing three major histologic groups that have prognostic significance.12 (1) Embryonal histology, which is favorable, accounts for 90% of genitourinary RMS. (2) Botryoid histology, which is often seen in bladder and vaginal RMS, is essentially a variant of E-RMS. (3) Alveolar histology accounts for the remaining 10% of GU-RMS and is associated with the worst outcomes.3 E-RMS may occur in a solid form arising in muscle groups, such as the trunk and extremities, or as sarcoma botryoides, a polypoid variety that occurs in hollow organs or body cavities, such as the bladder or vagina. There is a spindle cell, or leiomyomatous, variant seen frequently in the paratesticular region. The botryoid and spindle cell variants of E-RMS are associated with an excellent survival.
The pathologic diagnosis of RMS goes beyond basic histologic classification and is complimented by such techniques as immunohistochemistry (IHC) and cytogenetics. Specifically, IHC staining has been helpful in the diagnosis and prognosis of A-RMS. A-RMS demonstrates high levels of myogenin staining, whereas E- RMS is either negative or has only low-level staining.13 Subsequently, diffuse myogenin staining has been demonstrated as predictive of decreased survival independent of tumor stage, site or histology.14 Additionally, E-RMS and A-RMS have been noted to have distinct cytogenetic abnormalities. A-RMS is associated with a translocation between chromosomes 1 or 2 and 13, resulting in the formation of a chimeric protein. PAX3, a DNA-binding protein on chromosome 2, or PAX7, a DNA-binding protein and on chromosome 1, is fused to the FKHR gene on chromosome 13.15, 16 Expression of t(2;13), PAX3-FKHR is an adverse prognostic factor for children presenting with metastatic A-RMS. In addition, patients with the t(1;13) translocation, PAX7-FKHR fusion, are often younger and have a better prognosis than do their counterparts with the t(2;13), PAX3-FKHR fusion abnormality.16, 17 This has led some to propose a modified risk-stratification which includes this translocation status.18 However, analysis of a large series of cases consistently fails to show an absolute association of PAX-FHKR translocations with A-RMS; at least 25% of these tumors possess classic alveolar histology but lack a translocation. In contrast, E-RMS does not demonstrate recurrent chromosomal translocations. Instead, they show greater genomic instability and recurring allelic imbalances such as loss of heterozygosity (LOH) at chromosome 11p15.5. This is at a different location than the WT2 gene implicated in the development of some Wilms tumors.19 This region is the location of the IGF-2 gene. IGF-2 over-expression has been documented in both A-RMS and E-RMS. IGF-2 is a growth factor that can stimulate the growth of RMS tumor cells, and antibodies directed against IGF-2 can inhibit tumor growth.20 Amplification of the transcription factor N-MYC has been noted in RMS, and over-expression of N-MYC was associated with an adverse outcome in A-RMS.21 In summary, utilizing these advanced, adjunctive techniques to standard histopathology, classification based on gene expression is possible using as few as five genes with an estimated error rate of less than 5%.19 To that end, the next round of Children’s Oncology Group (COG) studies for RMS will likely use translocation fusion status as the superseding biologic risk factor rather than embryonal or alveolar histology, i.e. alveolar histology with no translocation fusion will be managed just as an E-RMS tumor.
Staging/Grouping/Risk Classification of GU-RMS
The approach to classifying patients with RMS can initially appear complicated. However, utilizing the important factors predictive of outcome these systems attempt to logically “stage” the patients pre-surgically, “group” them based on completeness of resection, and “risk” stratify them based on histology, stage, and group (Tables I, II, and III). The Tumor-Node-Metastasis (TNM) system segregates patients by favorable and unfavorable sites and is now used in an integrated IRSG Staging. Favorable GU sites include paratesticular and Vaginal/Cervical/Uterine. Unfavorable GU sites are bladder and prostate. The presence of distant metastases at diagnosis, involved regional lymph nodes (LNs), and large primary tumors (>5 cm) at unfavorable sites are relatively unfavorable prognostic signs.22 One important item to note when assessing individual patients is that the post-surgical grouping is dependent to a large extent on the completeness of surgical excision. As the treatment of GU-RMS has evolved, more patients with Bladder/Prostate RMS undergo biopsy-only at the initial surgical procedure, leaving gross residual disease. This results in the shifting of more patients from group I to group III. Therefore, theoretically equivalent tumors could end up in different groups, depending on the aggressiveness of the initial surgical resection. IRSG and current COG RMS studies group patients into low-, intermediate-, and high-risk groups. This risk-classification combines both the clinical grouping and TNM systems with the addition of histology. The system attempts to classify patients based on the various prognostic variables known to predict outcomes. One recent change to this system occurred with COG Study ARST0431 which included all ages of Stage IV E-RMS in the high-risk group. Formerly, Stage IV E-RMS patients under the age of 10 years-old were considered intermediate-risk.
General Treatment Paradigm
The treatment of GU-RMS has evolved away from radical surgical excision towards organ sparing approaches when possible. Once it was demonstrated that most patients would survive the disease, investigators explored the use of primary chemotherapy and radiation therapy to avoid the surgical exenteration generally employed for GU-RMS. The most recent studies have thus focused on further morbidity and therapy reduction. The current round of COG studies has recently concluded and results will be anticipated in the coming years. For low-risk patients with embryonal/botryoid/spindle-cell RMS, ARST0331 opened in 2004. This study evaluated reducing the duration of chemotherapy for low-stage, favorable-site tumors. The study for intermediate-risk patients, ARST0531, evaluated the role of earlier radiation.
While the trend has generally been towards therapy reduction, studies have occasionally indicated that therapy needs to be intensified. For example, while it appears that boys less than 10 years of age with paratesticular RMS can safely have their retroperitoneum staged with cross-sectional imaging, those ≥ 10 years-old are under-staged with imaging alone. Thus, a staging ipsilateral retroperitoneal lymph node dissection (RPLND) is mandated in that age group.23, 24 Likewise, therapeutic intensification is under investigation for those who experienced poor outcomes in prior studies. ARST0431, the study for high-risk patients, investigated more intensive chemotherapy regimens in an attempt to improve survival in this population.
Paratesticular RMS
Background
Paratesticular RMS (PT-RMS) makes up 5 to 10% of all new RMS diagnoses and occurs in a bimodal age distribution, with approximately 1/3rd being diagnosed between age 1 to 5 years-old and another 1/3rd diagnosed after 10 years of age.25, 26 PT-RMS arises in the muscle of the distal portion of the spermatic cord and may invade the testis or surrounding tissues. Presentation is often a unilateral painless scrotal swelling or mass that is distinct from the testis. Because of this superficial location and the ease with which it can be examined and noted, PT-RMS is usually detected and diagnosed earlier. For example, 60 to 80% of PT tumors are stage 1, compared with 10 to 15% of all primary RMS locations.26 More than 90% of PT-RMS cases have embryonal histology and a good prognosis. However, regardless of histology, it is considered a favorable location because even patients with alveolar PT-RMS have a better prognosis than other alveolar RMS primaries.27
Clinical Presentation and Initial Management
PT-RMS is typically noted by the patient or family as a painless scrotal mass which may or may not be distinct from the testicle. The clinical approach to this situation involves a thorough history and physical exam followed by serum tumor markers to evaluate for a primary testicular malignancy (AFP, bHCG, LDH) as well as a scrotal ultrasound to attempt to distinguish between a testicular or paratesticular primary location. Of note, the physical exam should specifically note any apparent invasion of the scrotal wall as well as any inguinal or cervical lymphadenopathy. Staging consists of serum electrolytes, complete blood count, liver function tests, and bilateral bone marrow aspirates/biopsies. Additional staging includes cross-sectional imaging of the chest, abdomen, and pelvis specifically investigating for the presence of enlarged retroperitoneal LNs or other metastatic sites (Figure 1).
Axial pelvic CT images of primary tumor in a boy with paratesticular RMS
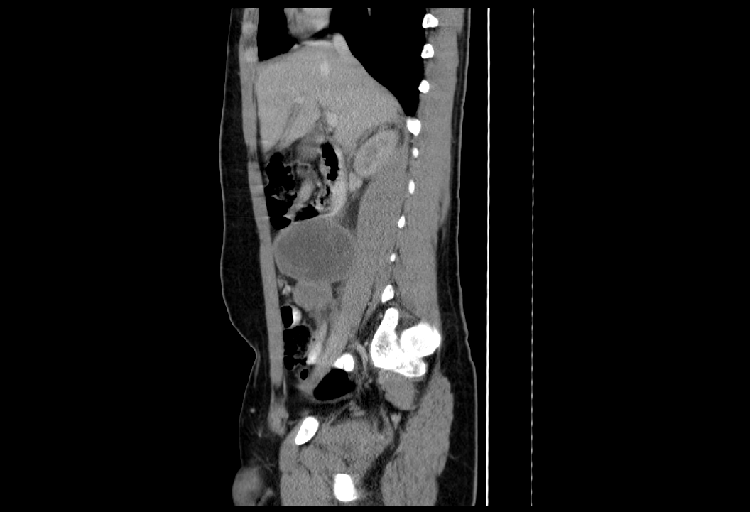
Sagittal abdominal CT images of massive lymphadenopathy in a boy with paratesticular RMS
Bone scans have traditionally been part of staging at diagnosis per the cooperative study group protocols; however, recent investigation suggests that FDG-PET scans can be of more benefit in staging and for future comparison during and after treatment to evaluate for therapeutic effectiveness (Figure 2).28, 29
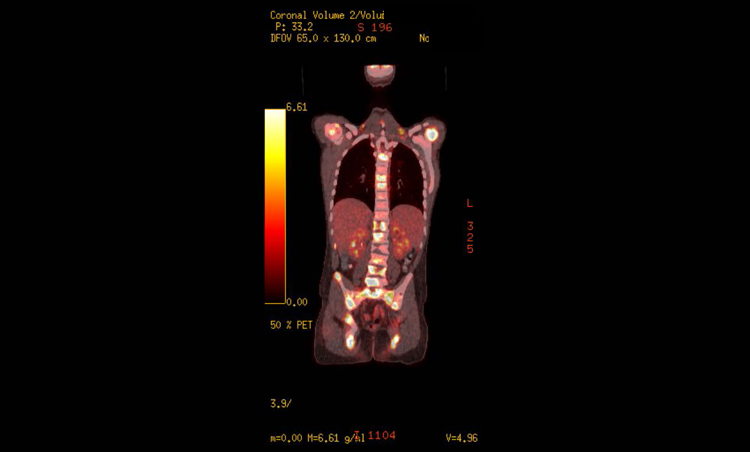
PET images of widespread bony and soft-tissue metastases in a boy with paratesticular RMS
Orchiectomy
Regardless of the presence or absence of metastatic disease on staging imaging, the next step in management is excision of the primary which should always be approached by radical inguinal orchiectomy. There is no role for scrotal exploration for excision and such surgery would then mandate a secondary excision of this scrotal scar as well as an inguinal incision to resect the remaining inguinal spermatic cord. Completeness of this resection will impact further management including radiation dosing.
Adjuvant Therapy
RPLND
Additional surgery at this point depends on the patient’s age and the initial staging imaging. Prior studies investigated forgoing staging ipsilateral RPLND; however, they demonstrated that those boys 10 years of age and older were at higher risk of being understaged by imaging alone than those under 10 years of age.23, 24, 30 Thus, current recommendations are to perform ipsilateral RPLND for all boys 10 years of age or older. The RPLND can be done in the traditional, open fashion, or via minimally-invasive techniques by surgeons experienced with such an approach.31, 32 Additionally, boys of any age who are found to have retroperitoneal metastases should receive RPLND. The exception to this treatment plan is in the presence of bulky lymph node involvement (more than 2 masses greater than 2cm). In the setting of such advanced disease on imaging, biopsy can confirm metastasis to allow for appropriate staging and prompt initiation of therapy. Completion RPLND after induction chemotherapy is indicated if the first cycles of chemotherapy have allowed for enough reduction that an RPLND could safely remove all gross disease. If this metastatic RMS is resected from the retroperitoneum, dose-adjusted radiotherapy is then administered. If these retroperitoneal metastases are not resected, radiation at 5040cGy is given to the residual masses and RPLND to resect this residual may again be considered at the completion of all chemotherapy and radiation if such masses are still radiographically apparent.
Chemotherapy and Radiation
Currently there are studies underway to assess chemotherapy reduction for embryonal PT-RMS patients because they generally represent a low-risk population. Similarly, there are studies investigating dose intensification for alveolar PT-RMS. However, these recommendations in this chapter follow the most current standards of care. Regardless of histology, PT-RMS patients who are either N0 or N1, by imaging if less than 10 years-old or by RPLND if 10 years of age or older, and have no evidence of non-nodal distant metastasis are treated with 45 weeks of Vincristine, Actinomycin-D, and Cyclophosphamide (VAC). Of note, RMS patients may often receive Cyclophosphamide or Ifosfamide and are at risk for hemorrhagic cystitis and other late bladder effects and are thus managed with hyperhydration and Mesna at the time of chemotherapy administration.
Stage 1 Group I patients (N0) do not require radiotherapy. Local Group II PT-RMS patients represent those who have had local spillage at the paratesticular site. These patients require radiotherapy to this local site, which would traditionally be 4140cGy. However, if bulk disease remains at the primary site, second-look surgery should be considered, which may include hemi-scrotectomy, in order to obtain resection of residual disease. This would allow dose reduction to 3600cGy to the PT site if complete resection is obtained. Additionally, just as any patient with PT-RMS, if Local Group II patients also have evidence of metastatic retroperitoneal LNs they will additionally require retroperitoneal radiotherapy. The dose of radiation to the retroperitoneum depends on the status of these nodes. If the LNs are grossly excised at RPLND and are positive (Retroperitoneal Group II), this dose is 4140cGy. However, if there is gross residual disease in the retroperitoneum (Group III) either by incomplete excision or biopsy-only, this dose is increased to 5040cGy.
For widely metastatic disease (i.e. not only in the retroperitoneum), the treatment paradigm is quite different. These patients are considered high-risk regardless of their histology. Formerly, the Stage 4 patients under the age of 10 years with E-RMS were considered to be intermediate-risk. On the current studies these patients are included in with all histologies and Stage 4 Group IV disease to be classified as high-risk. These patients are to undergo a radical inguinal orchiectomy for diagnosis followed by enrollment on study. All such patients now receive 51 weeks of intensive chemotherapy with VAC plus Irinotecan, Ifosfamide, Etoposide, and Doxorubicin. The radiotherapy plan depends on the presence and volume of metastatic disease in the retroperitoneum and the completeness of resection at the time of orchiectomy. As mentioned earlier, staging RPLND has a role in assessing retroperitoneal spread, except in those patients with bulky disease (more than 2 masses greater than 2cm). In the setting of more advanced disease on imaging, initial retroperitoneal mass biopsy can confirm metastasis and allow prompt risk-assessment and initiation of therapy. Completion RPLND after induction chemotherapy is indicated if the first 20 weeks of chemotherapy allow for enough disease reduction that RPLND could remove all gross disease. If this metastatic RMS is resected, radiotherapy is then administered. However, if these retroperitoneal metastases are not resected, radiation at 5040cGy is first given to these residual nodes. RPLND for resection of this residual may again be considered at the completion of the 51 weeks of chemotherapy if still such masses are radiographically apparent. All other non-nodal sites of metastasis are given 5040cGy of radiation.
As for the local control in the setting of Stage IV disease, this is similar to those with Stage I disease. That is, if there is spillage at the time of the orchiectomy and gross disease remains, a second operation which may include hemiscrotectomy to resect this disease can allow for a reduction in radiotherapy required at the local site. For patients with PT-RMS and completely resected disease at the primary paratesticular site from a radical inguinal orchiectomy with negative margins, no PT radiation is given. For those who have paratesticular microscopic or gross residual disease radiation is administered to the inguinal and scrotal fields.
Prognosis
The prognosis for patients with PT-RMS is generally excellent in the setting of localized disease (Stage 1 Group I-III).26 The best prognosis is seen in those with Stage 1 Group I and II disease where the overall survival (OS) and event-free survival (EFS) are 94 to 96% and 91 to 95%, respectively. Those with Group III disease have a slightly worse prognosis with 5-year EFS and OS of 75% and 76%, respectively. These outcomes appear to be somewhat independent of alveolar or embryonal histology.27 Patients with alveolar PT-RMS and localized disease (Stage 1 Group I-III) had a 5-year EFS and OS of 78% and 89%, respectively.27 However, it remains to be seen whether these alveolar PT-RMS patients are more likely to have the t(1;13) translocation, PAX7-FKHR fusion which may not be associated with poor outcomes more typical of the older patients with extremity A-RMS which generally have the t(2;13), PAX3-FKHR fusion abnormality. Regardless of such histopathologic risk-assessment, patients with metastatic Stage IV disease have a worse prognosis with a 5-year OS reported as approximately 20 to 25%.26
Bladder and/or Prostate
Epidemiology
Bladder and/or prostate (B/P) are the most common sites of GU-RMS, accounting for ~5% of all RMS. RMS is the most common bladder neoplasm in children under 10 years of age; the median age of children presenting with B/P RMS is 5 years.30
Clinical Presentation and Initial Evaluation
B/P RMS typically presents with hematuria, stranguria, urinary frequency, and/or acute urinary retention.33 As a general rule, a preschool-aged boy with acute urinary retention should be considered to be at high risk of RMS, and should be worked up accordingly. In girls with an intraluminal sarcoma botryoides RMS, tumor may be visibly prolapsing at the urethral meatus (Figure 3). Physical exam findings on two girls with sarcoma botryoides variant of RMS
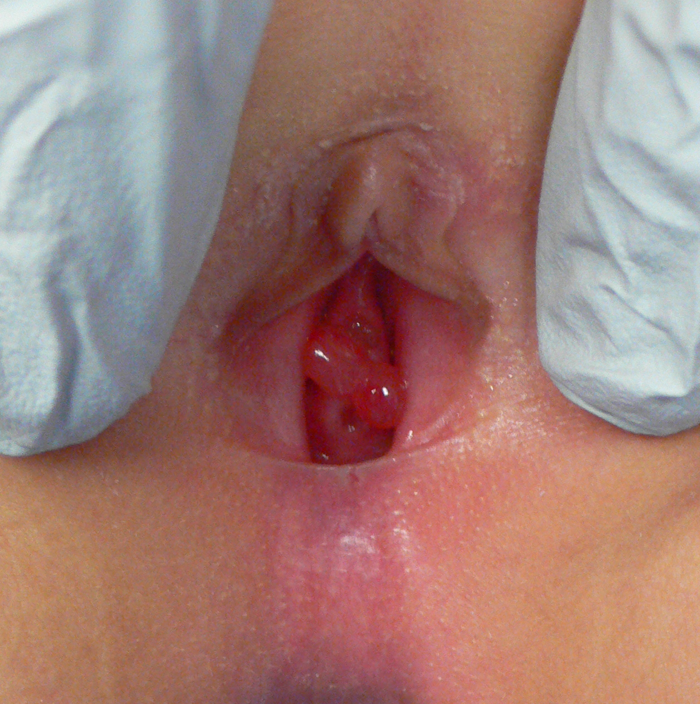
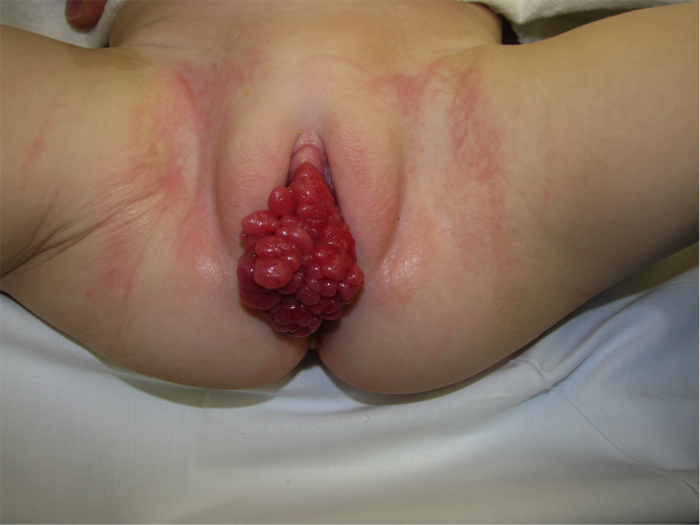
On physical exam, a palpable lower abdominal mass is often present, representing the mass itself, a distended bladder, or both. Prostatic RMS in particular is typically palpable on rectal examination.
Differentiating between bladder and prostate as the primary site of origin from which a large RMS arose can be quite challenging. In general, prostatic primaries are more likely to be large, solid masses. By contrast, bladder RMS primaries tend to occur as the sarcoma botryoides form (Figure 4);
these tumors are frequently located at the trigone or bladder neck.34
The initial evaluation of a child with suspected B/P RMS should consist of ultrasound if an abdominal mass is not palpable. If tumor or a distended bladder can be palpated, or if an initial ultrasound reveals a suspicious mass, then cross-sectional imaging with CT or MRI should be performed in order to determine the extent of local tumor (Figure 5). In addition, these modalities can evaluate the retroperitoneum for evidence of lymphatic or distant metastatic spread.
Treatment
Initial Surgical Management
Historically, the usual treatment of B/P RMS was surgical excision via radical cystoprostatectomy or, more commonly, pelvic exenteration.35 Beginning with the IRSG studies in the 1970s, however, chemotherapy and radiation began to supplant surgery as the mainstay of treatment. Indeed, a major goal of IRS-II (1978-1984) was to preserve a functional urinary tract. This focus has sharpened in more recent years.36 Currently, the major components of surgical management are an initial biopsy (ideally by endoscopic or percutaneous means), followed by chemotherapy and/or radiotherapy, and lastly, if needed and if feasible, exenterative surgery.
Diagnosis of B/P RMS obviously requires pathological review of an adequate sample of tumor tissue. Depending on the size and location of the tumor, this should be accomplished by minimally-invasive means such as transurethral resection (the preferred method if feasible), percutaneous needle biopsy, or ultrasound-guided transrectal or transperineal biopsy. Occasionally, these methods are not feasible, in which case an open biopsy may be necessary via a low midline or pfannenstiel incision. In such cases, an attempt at lymph node sampling (such as the iliac or obturator nodes) is recommended in order to improve staging accuracy.
In cases when the tumor can be completely excised with minimal morbidity – for example, in a patient with a small RMS at the dome of the bladder – then consideration can be given to partial cystectomy with wide local margins.37 A significant advantage to this approach is that, if completed with negative margins, the patient may be able to retain a relatively normal bladder. In IRS-IV, this strategy was successfully applied in a number of patients, with 13 of 17 children undergoing initial partial cystectomy having no evidence of disease at long-term follow-up. Radiation therapy was required in 10 of these patients, however, with implications for postoperative bladder function.36
If the tumor is sufficiently large to obstruct the ureter(s) and or bladder drainage, then temporary urinary diversion should be performed via a JJ ureteral stent, percutaneous nephrostomy tube, and/or Foley catheter as appropriate.33
Pretreatment Re-Excision (PRE)
In rare cases, pretreatment re-excision (PRE) – often by partial cystectomy, cystectomy, or cystoprostatectomy – may be considered if a complete excision is feasible. Although pretreatment re-excision may not infrequently be necessary in non-RMS tumors,38 this treatment is not typically employed in B/P RMS. That said, patients managed with an unplanned or incomplete excision in whom PRE can render the patient disease-free, are potentially candidates for this approach.
Second Look Operation (SLO)
Following chemotherapy and/or radiation therapy, residual masses may be present. A second-look procedure is frequently necessary in such cases, including but not limited to cystoscopy, biopsy, exploration, partial cystectomy, cystoprostatectomy, or prostatectomy. In IRS-IV, 53 of 88 B/P RMS patients ultimately required at least 1 second-look operation.36 Assessing the completeness of therapeutic response, however, can be challenging. Mature rhabdomyoblasts are not infrequently present in post-treatment biopsies, and these can be easily confused with active disease (particularly on frozen section biopsy). Although there is a generally low recurrence rate from mature rhabdomyoblasts,39 deaths have been reported following this diagnosis.40
Post-treatment Bladder Function
As noted above, B/P RMS patients are managed with intensive surgery, chemotherapy and/or radiation therapy. Radiation therapy, in particular, has a particularly important role in B/P RMS algorithms. In both IRS-IV and the SIOP MMT89 protocol, higher local recurrence rates were noted in patients who did not receive radiation.36, 41 However, OS rates were similar between those who did and did not receive radiation.41 However, radiation therapy can have a substantial impact on bladder function. In IRS-IV, 55% of event-free, surviving patients were reported to have “preserved” bladder function. However, this figure was determined based on patient questionnaire only, and only 1 child underwent formal urodynamics testing.36 In a smaller series reported by Yeung et al, all patients who received pelvic irradiation had markedly abnormal urodynamic profiles, specifically reduced functional capacity and atypical voiding curves.42 Among all children with B/P RMS in IRS-IV, only 40% were event-free with normal functioning bladders – and as noted above, this is likely an overestimate given the lack of formal urodynamic assessments in these children.36
Abnormalities in bladder function may arise secondary to surgical (e.g. nerve damage, partial cystectomy) or medical management.43-45 Typical presenting complaints include lower urinary tract symptoms, including enuresis; the latter may be more common in children with a history of genitourinary RMS than in survivors of other childhood cancers.42 Evaluation should include not only an assessment of symptom severity and bother, but also integrity of the upper tracts, as abnormal elimination habits (whether in the storage or voiding phase, or both) may place renal units already exposed to chemotherapy and/or radiation at risk.
Hemorrhagic cystitis, which often develops acutely in patients receiving chemotherapy with alkylating agents, may also present as a late effect, with a widely variable presentation.46 Radiation- and chemotherapy-induced cystitis is correlated histologically with local tissue hypoxia, ischemia, and necrosis. Cyclophosphamide and its metabolite acrolein are considered the classic etiologic agents, but other chemotherapeutic agents (e.g. dactinomycin and doxorubicin) may act synergistically to increase the risk of hemorrhagic cystitis.47-49 Furthermore, agents acting to decrease the toxicity of chemotherapeutic agents (e.g. MESNA, N-acetylcysteine) do not entirely eliminate the risk of hemorrhagic cystitis. Patients receiving pelvic radiation appear to be at increased risk of hemorrhagic cystitis, particularly when doses exceed 3000cGy for whole bladder exposure and 6000cGy for partial bladder exposure.50, 51 Doses exceeding 4500cGy are associated with more consistent development of radiation cystitis.51, 52 Even when the acute presentation is successfully managed (e.g. bleeding is stopped), hemorrhagic cystitis, decreased bladder compliance may result in late effects including urinary symptoms and altered lower tract function.53
Urinary diversion is not infrequently required in B/P RMS, either due to initial or delayed cystectomy or to radiation-related loss of bladder function. However, good quality of life can be restored to these patients.54 Many authors advocate for the use of initial incontinent urinary diversion, either by ileal or colonic conduits,55 while others have reported good outcomes following an immediate orthotopic continent diversion.37 Initial continent urinary diversion bears a particular set of challenges, however: first, any postoperative complication is likely to delay chemotherapy, and continent diversion is generally accepted to have a higher complication rate than incontinent diversions56; second, as noted above frozen section is unreliable at assessing the adequacy of surgical margins37, 40 ; third, if local recurrence does occur, a continent diversion makes adequate treatment quite challenging57; and fourth, given the age range in which B/P RMS occurs, it can be difficult to gauge whether the child and/or family are capable of long-term, adequate maintenance of their urinary diversion. Because of these specific challenges, many authors recommend against immediate reconstruction unless further therapy is highly unlikely and both the treating center and surgeon have extensive experience with continent reconstructions.
Patients with intestinal substitutions (neobladder, conduit, or augmentation cystoplasty) are at increased risk for metabolic abnormalities (hypochloremic, hypokalemic metabolic acidosis with ileal and colon conduits), calculi, and perforation. Use of bowel in the genitourinary tract is also associated with an increased risk of development of secondary malignancies, with the risk typically increasing between 5-10 years after surgery.58 Patients with urinary diversions should be assessed at least annually with upper tract imaging and labs (electrolytes, BUN, and creatinine); given the time course for development of secondary malignancies in this population, most urologists initiate annual cystoscopy and urine cytology 5-7 years after diversion. Since treatment for RMS has increasingly focused on organ preservation, urinary diversions are rarely employed at present.
Prognosis
Unlike other GU sites, B/P is considered to be an unfavorable site for RMS. As such, B/P thus carries a worse prognosis than other sites and under the IRSG staging system is – at best – considered to be Stage 2 before treatment. The EFS of B/P RMS in the IRS-IV trial was 77%; patients with non-metastatic disease had an OS of 82% at 6 years. However, there are groups with particularly good prognoses after a diagnosis of B/P RMS; children with an endophytic sarcoma botryoides tumor have been reported to have 10-year survival rates of >90%.40
Vulvar/Vaginal/Uterine
Epidemiology
Gynecologic malignancies are rare in children, and account for fewer than 1 in 20 childhood tumors, and Most of these are vaginal RMS.59, 60 Although RMS in gynecologic sites accounts for about 3.5% of all pediatric RMS,61 these tumors are still extremely rare: Fernandez reported 18 patients in 39 years at a single institution,62 while Kirsch found only 67 cases of female pediatric genitourinary RMS in 33 years.63 While gynecologic RMS can occur at any age, vaginal tumors are most common in toddlers and pre-school age children (with diagnosis occurring at a mean age of 3.7 years in one study).62 There is a bimodal distribution of female patients with RMS, with the first peak between 1 to 4 years old (mostly vaginal tumors) and another between 15 to 19 years old (mostly uterine/cervical primary sites).63 Most patients (81%) are Caucasian, with a minority being African American (13%) or Hispanic (2%).61 Overall, the vagina and uterus/cervix are the most common primary sites, although the exact proportions vary by study. Kirsch et al reported that the proportions were relatively equal (42% vagina compared with 39% uterus/cervix), although a review of IRS I-IV found the vagina as the primary site in 54% of patients, compared with 17% for the uterus and 15% for the cervix.61, 63 Given that these tumors are relatively uncommon, sampling error could account for these disparate observations.
As in other sites, the histology of gynecologic RMS may be either embryonal or alveolar. Uterine and vaginal tumors tend to be Group III or IV at presentation in most cases given the typical approach with an initial biopsy-only, compared with vulvar tumors, which are typically Group I or II because of the ability to offer upfront resection. The vast majority (50-67%) of embryonal vaginal tumors are localized at diagnosis; in those that are metastatic, the lung is most common site of distant disease.64 Arndt reported 45% alveolar histology in vulvar tumors.61 Uterine tumors tended to be large and invasive (>5 cm in most cases) and were associated with nodal disease in 27%. In contrast, only one patient with an invasive cervical RMS was identified in the IRS series,61 and cervical RMS has, to date, never been associated with nodal disease.
Tumor Biology
RMS is a small round blue cell tumor with differentiated skeletal muscle.65 Vaginal tumors most typically have two subtypes: botyroid (“bunch of grapes”) and embryonal. The botyroid appearance arises secondary to tumor growth in the subepithelium of hollow organs, with resulting distortion of the submucosal layer of spindle cells.66 The alveolar variant of RMS is uncommon in vaginal/vulvar/uterine sites, but portends poorer outcome except when associated with vulvar lesions (which tend to be localized).67
Clinical Presentation and Work Up
Diagnosis typically occurs after patients present with symptoms, rather than as incidental findings or prompted by screening. Vaginal and uterine RMS most commonly present with bloody vaginal discharge (67%) or a visible mass extending from the vagina (33%; Figure 3).59,62,68 The differential diagnosis of vulvar masses also includes other vulvar malignancies (e.g. endodermal sinus tumor) or benign vulvar growths (e.g. Gartner duct cysts, canal of Nuck cysts, and hamartoma).69 In patients in whom the vulvar mass is not readily identified as neoplastic, a karyotype should be considered, although of course this should not delay diagnosis or treatment in patients in whom neoplasia is suspected. Initial imaging may consist of a pelvic ultrasound in order to delineate the size and extent of the mass; if a neoplastic process is suspected, then cross sectional imaging of the abdomen and pelvis with either a CT or MRI will help to determine the extent of local and nodal disease. Since the lungs and bone marrow are the most common sites of metastatic disease, initial metastatic workup consists of a chest CT, bone marrow biopsy, and bone scan.
Treatment
Diagnosis
Historically (until 1972), treatment consisted of pelvic exenteration, but IRS I/II protocols (1972 and 1984, respectively) found that the vast majority of tumors were exquisitely sensitive to chemotherapy.70 Following the IRS I/II protocols, only 12% of patients required pelvic exenteration, meaning that uterine preservation could be achieved in 88% of patients;71 on IRS III/IV, only 22% of patients underwent hysterectomy and 82% were alive 5 years after diagnosis.61 Concurrently, a SIOP protocol open between 1984-1994 found a survival rate greater than 90% using multimodal therapy, and a uterine preservation rate of 88% among survivors (with two deaths and three patients requiring hysterectomy).72 Based on these findings, present recommendations are for upfront chemotherapy followed by surgical excision and in some cases adjuvant radiotherapy.71 Pelvic exenteration is thus reserved for patients with tumors refractory to chemotherapy and radiation.
Following chemotherapy, exploration is performed with complete tumor excision (when possible) and possibly another biopsy to assess tumor viability.61, 68, 73, 74 Local control is paramount as local recurrence accounts for the vast majority of treatment failures. A 5 mm margin is considered sufficient for oncologic control.74 While complete resection should be performed when possible (e.g. vaginal tumors may require hysterectomy), tumors extending into the bladder or bowel should be completely resected only when organ function can be preserved.74
In addition to radiographic evaluation, lymph nodes should also be assessed pathologically. Vulvar lesions drain to superficial and deep inguinal lymph nodes, while vaginal and uterine tumors drain to the pelvic lymph nodes. Surgical complications are discussed in more detail below, and include rectovaginal or vesicovaginal fistula and urinary incontinence, typically in patients with extensive resections including pelvic exenteration.62
Pretreatment Re-Excision (PRE)
Pre-treatment re-excision is typically employed for patients in whom a mass is “completely” excised (e.g. not biopsied) prior to the diagnosis of RMS. Histologic evaluation of RMS in these cases often will reveal positive margins and incomplete resection of the mass. Prior to administering adjuvant therapy, re-excision of the mass with removal of any suspected residual tissue with a wide local margin is appropriate. Lymph node sampling, if not performed at the time of initial surgery, should also be performed at this time. As with excision following chemotherapy and radiotherapy, excision of organs, such as the bladder, which may result in significant functional consequences to the patient, should be avoided.74
Second Look Operation (SLO)
Second look operations are periodically undertaken to assess tumor burden and clinical response following radiotherapy and chemotherapy; tumor excision may be performed if it will result in a decrease in additional therapy (e.g. radiation). Because vaginal and vulvar RMS are typically highly responsive to chemotherapy, SLOs are rarely performed for tumors in these sites.74
Adjuvant Treatment
Chemotherapy/Radiation
Standard chemotherapy for RMS is VAC. Patients who did not receive Adriamcyin had a 15% recurrence rate, compared with complete cure in patients given Adriamycin. Ifosfamide is occasionally substituted for cyclophosphamide. Radiation is used for patients in whom there is residual tumor following chemotherapy. Historically, the dose has been 5040cGy for patients with gross disease and 4140cGy for patients with microscopic disease. A recent COG study (D9602) set out to determine whether comparable survival results to those seen in IRS III/IV could be achieved with lower external beam radiotherapy doses (3600cGy in patients with microscopic positive margins, 4140cGy in patients with nodal disease, and 5040cGy in patients with gross residual disease).74 Girls with vaginal tumors accounted for the majority of the treatment failures, with local control achieved in only half of this subgroup, although many did not receive alkylating agents or radiotherapy, making the reason for failure challenging to determine with certainty. Current COG protocols mandate radiation therapy for group II and III vaginal tumors, with the intensity of radiotherapy ranging from 3600-5040cGy depending on the extent of residual tumor. In these patients, the local failure rate is increased without radiation.75
Radiation failure appears to be most pronounced in children under 3 years of age, possibly because of inadequate dosing related to the increased fear of radiation-related late complications as well as decreased comprehension of and cooperation with the goals of therapy.76, 77 Nonetheless, radiation failure appears to unequivocally increase the risk for recurrence (70%) and death. Although not routinely used in the United States, brachytherapy for pediatric vaginal and vulvar RMS has been employed in Europe.78, 79 Magne et al. reported a high (91%) five-year survival in 32 patients who received brachytherapy for residual disease, with only two experiencing acute radiation-induced side effects. However, the vaginal stenosis rate in this group was 20% over 8 years.
Prognosis and Follow Up
Factors portending an improved survival rate include young age (<10 years), locally confined disease, and chemotherapeutic treatment with VAC and (when appropriate) radiation.61 The vulva and vagina are considered favorable sites. Tumors larger than 5 cm, or those associated with local invasion or positive lymph nodes, were associated with poorer outcomes. Children older than one year had a better prognosis than infants, likely secondary to the ability to tolerate therapy-related complications.3, 80 Almost all deaths occurred in first 5 years after diagnosis, but late relapse has been reported.81
Late Effects
As RMS treatment centers on organ preservation, late effects increasingly focus on the sequelae of chemotherapy and radiotherapy rather than surgery. Spunt et al. observed multiple complaints in patients treated for RMS, including reproductive issues (dyspareunia, vaginal stenosis, vaginal fistula, endocrine effects, abnormal menstrual cycles, premature menopause), genitourinary conditions (abnormal bladder function, ureteral obstruction, pyelonephritis, urethral obstruction), and altered gastrointestinal function (strictures, fistula, perforation, cholecystitis, gastritis, fecal incontinence).82 Although this list is extensive, the authors cautioned that the late development of many complications (particularly in patients receiving radiation) may mean that the true prevalence of late effects is underestimated. In addition, at present there are no standardized methods to assess pelvic floor integrity and function in survivors of RMS, and so subtle bladder and bowel complaints may go unreported.
While the endocrine gonadal function is most commonly affected (e.g. premature ovarian failure, especially with pelvic radiation), the most common complications requiring surgical intervention were gynecologic complaints such as strictures and fistulae. Fistulae were universally associated with radiation therapy, with an equal proportion developing spontaneously and post-surgically. Vaginal stenosis can typically be managed with dilatation or more formal surgical repair; the risks of the latter include bowel perforation and recurrent stenosis, and so repair is typically undertaken only when conservative management has failed or has an extremely low likelihood of initial success.
Other systems in which late effects have been observed include the musculoskeletal system (scoliosis and tissue fibrosis), mental health issues (depression, anxiety, insomnia, academic issues, substance abuse), and cardiovascular (stroke, hypertension, doxorubicin-associated cardiomyopathy). Secondary neoplasms have a cumulative incidence of 13.5% at 20 years; Spunt et al. reported on three patients, all of whom developed secondary malignancies (osteosarcoma, squamous cell carcinoma in situ of the cervix, and colonic carcinoma) within the RMS radiation field. In this series, the proportion of patients developing secondary neoplasms was higher than that for other groups of cancer survivors, although it was unclear whether this finding was spurious given the small number of affected patients, or representative of a truly increased risk given the near-universal receipt of radiotherapy in that cohort. The likelihood of developing a late effect was not correlated with the age at diagnosis of RMS. The authors did find that refinements in therapy for RMS resulted in more late effects for patients treated after 1984 than those before, but this was due only to an increase in the number of lower grade (1 and 2) toxicity.
Radiation therapy is perhaps the single most important risk factor for the development of late complications. In the aforementioned study of 26 patients by Spunt et al., only two had no late effects, both of whom were radiation-naïve. Affected patients had an average of 4.5 late effects, with a maximum of 14 per patient; patients with radiation had a median of 9.5 late effects apiece compared with one late effect in patients who were radiation-naïve. The late effects were also more severe in patients who had undergone radiotherapy, with more severe (grade 3 to 4) effects reported in this group. Over half (54%) underwent surgery to manage complications, with vaginal dilatation being most common, followed by procedures to correct gastrointestinal strictures and vaginal fistulae. The authors concluded that radiation conferred a higher risk of treatment-related late complications in children than in adults. Given the close anatomic relationship in the pelvis between the gynecologic structures and the bladder, many of the same bladder late effects can be seen as in B/P RMS which were reviewed earlier.
Infertility
While fertility issues have been reported after treatment for many childhood cancers, the literature specific to fertility issues following treatment for vaginal/vulvar/uterine RMS remains scarce. Fertility may be challenged due to chemotherapy exposure, radiation therapy, and surgical intervention. Premature ovarian failure, as already discussed, is a common finding following pelvic radiation. In patients who have undergone hysterectomy, the ability to carry a child is lost; however, even in those in whom all or part of the uterus is preserved, post-surgical or post-radiation changes may promote cervical incompetence or premature labor. Live births have been reported after treatment for pelvic RMS, however.83 Pregnancy rates following treatment for sarcoma are moderate (47%) in at least one other study,84 suggesting that the pelvic radiation and surgical sequelae are stronger predictors of infertility than chemotherapy in girls.
In males who have received chemotherapy, infertility issues have been reported previously. One study described a 24% paternity rate, with the majority of other patients having oligo- or azoospermia.84 Patients with successful paternity did not have exposure to alkylating chemptherapy or pelvic radiation. Another study found universally abnormal semen analyses in males exposed to cyclophosphamide prior to puberty.85 The degree of oligo- or azoospermia was not dose-dependent in this study. While most patients did not have abnormalities of the hypothalamic-pituitary-gonadal axis associated with the abnormal semen analyses, 40% of boys had elevated luteinizing hormone levels despite normal testosterone, suggesting abnormal Leydig cell function. It is possible that ifosfamide may be less likely to cause significant fertility issues than cyclophosphamide.86, 87
Rare Primaries
Undistinguishable Pelvic
Pelvic RMS not arising from one of the genitourinary organs is rare. At presentation, these tumors are often large and extensive, encasing pelvic structures such as nerves and blood vessels. Just as with RMS clearly arising from a discrete organ, treatment is aimed at optimizing the chance for complete excision while minimizing damage to other organs and pelvic structures.88
Renal
Renal RMS is rare and in a 33 year period, the multicenter IRS group enrolled only 10 renal sarcomas and specifically only six cases of renal primary RMS.89 Therefore, prompt and accurate diagnosis requires a high index of suspicion and may be challenging. Both Wilms Tumor and renal cell carcinoma should be excluded from the differential diagnosis. Myogenin and MyoD1 are skeletal muscle markers which are considered both sensitive and specific for the diagnosis of RMS as both are expressed early in skeletal muscle differentiation.90 While WT1 positivity may be seen, typically a nuclear staining pattern is not seen in RMS as it is in WT.91 Desmin is also a marker for RMS.92
A retrospective review of renal primary RMS from the IRSG demonstrated that tumors were generally large (median diameter ranged from 8 to 15 cm), although the majority were noninvasive. Renal RMS had a high incidence of anaplasia (67%), much higher than reported in other childhood renal tumors or in RMS overall. One of the six patients had nodal disease and two patients presented with metastases. In this series, all tumors were of the embryonal subtype.89
As with RMS in other primary locations, the extent of excision of renal RMS appears to correlate with survival. COG recommends complete primary excision of the tumor when possible, including regional lymph node dissection for disease staging. Renal RMS is considered an unfavorable site. In the 6 patient IRSG series, initial treatment consisted of radical nephrectomy followed by 3 drug chemotherapy (VAC) in most cases, with radiation reserved for patients with residual disease. Three of six patients had recurrent disease in the brain or bone, and died of disease. The other three were alive and NED more than a decade after completing therapy.89 At present, biopsy followed by upfront chemotherapy is advocated to facilitate tumor removal and maximally preserve both affected and adjacent organs at the time of surgery. Following chemotherapy, radiation therapy can be employed in patients with residual tumor (positive margins or nodal disease) if the benefits outweigh the risks, even in the presence of abnormal abdominal anatomy.93 However, in the IRSG series, even with adjuvant radiotherapy, patients with nodal disease or metastases fared poorly.
Retroperitoneal
Retroperitoneal RMS as a primary site is rare; together, retroperitoneal and non-genitourinary pelvic RMS account for only 10% of all RMS,94 and in one institutional series, only seven retroperitoneal-primary RMS were recorded in a 30 year period.95 Tumors are typically identified after the patient develops symptoms (often nonspecific, such as pain or abdominal discomfort), and therefore many tumors are quite large (most are greater than 5 cm in diameter at the time of diagnosis). Retroperitoneal-primary RMS tumors also tend to be more locally extensive; Pham et al. reported that only about one-third of patients had completely localized disease: 62% had tumors affecting nearby organs, and although none had distant metastases at diagnosis, 14% were found to have metastatic disease over the course of treatment.95 Blakely et al. observed that 94% of tumors were clinical group III or IV, and in children in whom invasion status was recorded, 96% had invasive tumors.94
Most tumors are at the thoracolumbar level, with a significant minority at the lumbosacral level.95 Boys and children younger than 5 years were more commonly affected; in these groups, embryonal histology predominated, although alveolar histology was more common in older (e.g. adolescent) children. Improved failure-free and overall survival was predicted by younger age at diagnosis, female gender, and embryonal histology. Patients with embryonal histology were more likely to undergo tumor debulking compared with children with alveolar histology (who typically underwent biopsy alone); surgical debulking, even when the tumor was not completely resected, was correlated with improved survival. While patients with RMS had substantially shorter survival than patients with other types of sarcoma, the overall five-year survival was similar in patients with and without RMS, although this finding might reflect the small number of patients included.
Table I - Pre-operative TNM and IRSG Staging
TNM Staging
- T-stage
|
-
|
- T1
|
- Confined to anatomic site of origin
|
- T1a
|
- ≤5cm in diameter in size
|
- T1b
|
- >5cm in diameter in size
|
- T2
|
- Extension and/or fixed to surrounding tissue
|
- T2a
|
- ≤5cm in diameter in size
|
- T2b
|
- >5cm in diameter in size
|
-
|
-
|
- N-stage
|
-
|
- N0
|
Regional nodes not clinically involved |
- N1
|
- Regional nodes clinically involved by neoplasm
|
- Nx
|
- Clinical status of regional nodes unknown (especially sites that preclude evaluation)
|
-
|
-
|
- M-stage
|
-
|
- M0
|
- No distant metastasis
|
- M1
|
- Metastasis Present
|
-
-
- IRSG Staging
- Stage
|
- Site
|
- T
|
- N
|
- M
|
- 1
|
- Vaginal/Uterine/Cervical and Paratesticular
|
- Any
|
- Any
|
- M0
|
- 2
|
- Bladder/Prostate
|
- T1a/T2a (≤5cm)
|
- N0 or Nx
|
- M0
|
- 3
|
- Bladder/Prostate
|
- T1a/T2a (≤5cm)
- OR
- T1b/T2b (>5cm)
|
- N1
-
- Any
|
- M0
-
- M0
|
- 4
|
- Any
|
- Any
|
- Any
|
- M1
|
Table II – Post-Surgical Clinical Grouping
- Group
|
-
|
- I
|
- Localized disease, completely resected (microscopically negative margins), and regional nodes not involved
- Lymph node biopsy or sampling is highly advised
|
- Ia
|
- Confined to organ of origin
|
- Ib
|
- Contiguous involvement – infiltration outside the muscle or organ of origin, as through fascial planes
|
-
|
-
|
- II
|
- Total gross resection with evidence of regional spread
|
- IIa
|
- Grossly resected tumor with microscopic residual disease, but no regional nodal involvement
|
- IIb
|
- Regional disease with involved nodes, but completely resected with no microscopic residual
|
- IIc
|
- Regional disease with involved nodes, grossly resected, but with evidence of microscopic residual and/or histologic involvement of the most distal regional node in the dissection.
|
-
|
|
- III
|
- Incomplete resection with gross residual disease
|
- IIIa
|
- After biopsy only
|
- IIIb
|
- After gross major resection of the primary (>50%)
|
-
|
-
|
- IV
|
- Distant Metastatic disease present at onset
|
Table III – Risk Classification
General
Risk |
Histology |
Post-Op Group |
Pre-Op IRSG Stage |
Age |
Low |
Embryonal |
I, II, III |
1 |
All |
|
Embryonal |
I, II |
2, 3 |
All |
|
|
|
|
|
Intermediate |
Embryonal |
III |
2, 3 |
All |
|
Alveolar |
I, II, III |
1, 2, 3 |
All |
|
|
|
|
|
High |
Embryonal |
IV |
4 |
All |
|
Alveolar |
IV |
4 |
All |
GU Site Specific Risk Classifications
Primary Site |
Pre-Op Stage |
Initial Surgical Approach |
Post-Op Group |
Histology |
Age |
Risk |
Paratesticular |
1 |
Orchiectomy |
I, II, III |
Embryonal |
All |
Low |
|
1 |
Orchiectomy |
I, II, III |
Alveolar |
All |
Intermediate |
|
4 |
Orchiectomy |
IV |
Embryonal |
All |
High |
|
4 |
Orchiectomy |
IV |
Alveolar |
All |
High |
|
|
|
|
|
|
|
Vaginal/Uterine/
Cervical |
1 |
Resection |
I, II, III |
Embryonal |
All |
Low |
|
1 |
Biopsy |
III |
Embryonal |
All |
Low |
|
1 |
Resection |
I, II, III |
Alveolar |
All |
Intermediate |
|
1 |
Biopsy |
III |
Alveolar |
All |
Intermediate |
|
4 |
Resection or Biopsy |
IV |
Embryonal |
All |
High |
|
4 |
Resection or Biopsy |
IV |
Alveolar |
All |
High |
|
|
|
|
|
|
|
Bladder/Prostate |
|
|
|
|
|
|
|
2 or 3 |
Resection |
I or II |
Embryonal |
All |
Low |
|
2 or 3 |
Resection |
III |
Embryonal |
All |
Intermediate |
|
2 or 3 |
Biopsy |
III |
Embryonal |
All |
Intermediate |
|
2 or 3 |
Resection |
I, II, III |
Alveolar |
All |
Intermediate |
|
2 or 3 |
Biopsy |
III |
Alveolar |
All |
Intermediate |
|
4 |
Resection or Biopsy |
IV |
Embryonal |
All |
High |
|
4 |
Resection or Biopsy |
IV |
Alveolar |
All |
High |
Key of Abbreviations:
Rhabdomyosarcoma (RMS)
Embryonal Rhadomyosarcoma (E-RMS)
Botryoid Rhabdomyosarcoma (B-RMS)
Alveolar Rhabdomyosarcoma (A-RMS)
Bladder and/or prostate (B/P)
Genitourinary (GU)
Immunohistochemistry (IHC)
Loss of Heterozygosity (LOH)
Intergroup Rhabdomyosarcoma Study Group (IRSG)
Children’s Oncology Group (COG)
Retroperitoneal lymph node dissection (RPLND)
Paratesticular (PT)
Vincristine, Actinomycin-D, and Cyclophosphamide (VAC)
Tumor-Node-Metastasis (TNM)
Lymph Nodes (LNs)
Overall survival (OS)
Event-free survival (EFS)
References:
1. Dasgupta, R., Rodeberg, D. A.: Update on rhabdomyosarcoma. Semin Pediatr Surg, 21: 68, 2012
2. Crist, W., Gehan, E. A., Ragab, A. H. et al.: The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol, 13: 610, 1995
3. Meza, J. L., Anderson, J., Pappo, A. S. et al.: Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children's Oncology Group. J Clin Oncol, 24: 3844, 2006
4. Pappo, A. S., Shapiro, D. N.: Rhabdomyosarcoma: biology and therapy. Cancer Treat Res, 92: 309, 1997
5. Perez, E. A., Kassira, N., Cheung, M. C. et al.: Rhabdomyosarcoma in children: a SEER population based study. J Surg Res, 170: e243, 2011
6. Ferrer, F. A., Isakoff, M., Koyle, M. A.: Bladder/prostate rhabdomyosarcoma: past, present and future. J Urol, 176: 1283, 2006
7. Joshi, D., Anderson, J. R., Paidas, C. et al.: Age is an independent prognostic factor in rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. Pediatr Blood Cancer, 42: 64, 2004
8. Malempati, S., Hawkins, D. S.: Rhabdomyosarcoma: review of the Children's Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer, 59: 5, 2012
9. Sung, L., Anderson, J. R., Arndt, C. et al.: Neurofibromatosis in children with Rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma study IV. J Pediatr, 144: 666, 2004
10. Malkin, D., Li, F. P., Strong, L. C. et al.: Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science, 250: 1233, 1990
11. Ruymann, F. B., Maddux, H. R., Ragab, A. et al.: Congenital anomalies associated with rhabdomyosarcoma: an autopsy study of 115 cases. A report from the Intergroup Rhabdomyosarcoma Study Committee (representing the Children's Cancer Study Group, the Pediatric Oncology Group, the United Kingdom Children's Cancer Study Group, and the Pediatric Intergroup Statistical Center). Med Pediatr Oncol, 16: 33, 1988
12. Asmar, L., Gehan, E. A., Newton, W. A. et al.: Agreement among and within groups of pathologists in the classification of rhabdomyosarcoma and related childhood sarcomas. Report of an international study of four pathology classifications. Cancer, 74: 2579, 1994
13. Dias, P., Chen, B., Dilday, B. et al.: Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol, 156: 399, 2000
14. Heerema-McKenney, A., Wijnaendts, L. C., Pulliam, J. F. et al.: Diffuse myogenin expression by immunohistochemistry is an independent marker of poor survival in pediatric rhabdomyosarcoma: a tissue microarray study of 71 primary tumors including correlation with molecular phenotype. Am J Surg Pathol, 32: 1513, 2008
15. Gordon, T., McManus, A., Anderson, J. et al.: Cytogenetic abnormalities in 42 rhabdomyosarcoma: a United Kingdom Cancer Cytogenetics Group Study. Med Pediatr Oncol, 36: 259, 2001
16. Anderson, J., Gordon, T., McManus, A. et al.: Detection of the PAX3-FKHR fusion gene in paediatric rhabdomyosarcoma: a reproducible predictor of outcome? Br J Cancer, 85: 831, 2001
17. Sorensen, P. H., Lynch, J. C., Qualman, S. J. et al.: PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children's oncology group. J Clin Oncol, 20: 2672, 2002
18. Missiaglia, E., Williamson, D., Chisholm, J. et al.: PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol, 30: 1670, 2012
19. Davicioni, E., Anderson, M. J., Finckenstein, F. G. et al.: Molecular classification of rhabdomyosarcoma--genotypic and phenotypic determinants of diagnosis: a report from the Children's Oncology Group. Am J Pathol, 174: 550, 2009
20. El-Badry, O. M., Minniti, C., Kohn, E. C. et al.: Insulin-like growth factor II acts as an autocrine growth and motility factor in human rhabdomyosarcoma tumors. Cell Growth Differ, 1: 325, 1990
21. Williamson, D., Lu, Y. J., Gordon, T. et al.: Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J Clin Oncol, 23: 880, 2005
22. Lawrence, W., Jr., Anderson, J. R., Gehan, E. A. et al.: Pretreatment TNM staging of childhood rhabdomyosarcoma: a report of the Intergroup Rhabdomyosarcoma Study Group. Children's Cancer Study Group. Pediatric Oncology Group. Cancer, 80: 1165, 1997
23. Stewart, R. J., Martelli, H., Oberlin, O. et al.: Treatment of children with nonmetastatic paratesticular rhabdomyosarcoma: results of the Malignant Mesenchymal Tumors studies (MMT 84 and MMT 89) of the International Society of Pediatric Oncology. J Clin Oncol, 21: 793, 2003
24. Wiener, E. S., Anderson, J. R., Ojimba, J. I. et al.: Controversies in the management of paratesticular rhabdomyosarcoma: is staging retroperitoneal lymph node dissection necessary for adolescents with resected paratesticular rhabdomyosarcoma? Semin Pediatr Surg, 10: 146, 2001
25. Wiener, E. S., Lawrence, W., Hays, D. et al.: Retroperitoneal node biopsy in paratesticular rhabdomyosarcoma. J Pediatr Surg, 29: 171, 1994
26. Ferrari, A., Bisogno, G., Casanova, M. et al.: Paratesticular rhabdomyosarcoma: report from the Italian and German Cooperative Group. J Clin Oncol, 20: 449, 2002
27. Ferrari, A., Bisogno, G., Casanova, M. et al.: Is alveolar histotype a prognostic factor in paratesticular rhabdomyosarcoma? The experience of Italian and German Soft Tissue Sarcoma Cooperative Group. Pediatr Blood Cancer, 42: 134, 2004
28. Federico, S. M., Spunt, S. L., Krasin, M. J. et al.: Comparison of PET-CT and conventional imaging in staging pediatric rhabdomyosarcoma. Pediatr Blood Cancer, 60: 1128, 2013
29. Burnette, J. O., Klaassen, Z., Hatley, R. M. et al.: Staging Paratesticular Rhabdomyosarcoma in the "as Low as Reasonably Achievable" Age: The Case for PET-CT. Urology, 2013
30. Crist, W. M., Anderson, J. R., Meza, J. L. et al.: Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol, 19: 3091, 2001
31. Tomaszewski, J. J., Sweeney, D. D., Kavoussi, L. R. et al.: Laparoscopic retroperitoneal lymph node dissection for high-risk pediatric patients with paratesticular rhabdomyosarcoma. J Endourol, 24: 31, 2010
32. Cost, N. G., Dajusta, D. G., Granberg, C. F. et al.: Robot-Assisted Laparoscopic Retroperitoneal Lymph Node Dissection in an Adolescent Population. J Endourol, 2012
33. Meir, D. B., Inoue, M., Gur, U. et al.: Urinary diversion in children with pelvic tumors. J Pediatr Surg, 39: 1787, 2004
34. Hays, D. M., Raney, R. B., Jr., Lawrence, W., Jr. et al.: Bladder and prostatic tumors in the intergroup Rhabdomyosarcoma study (IRS-I): results of therapy. Cancer, 50: 1472, 1982
35. Lerner, S. P., Hayani, A., O'Hollaren, P. et al.: The role of surgery in the management of pediatric pelvic rhabdomyosarcoma. J Urol, 154: 540, 1995
36. Arndt, C., Rodeberg, D., Breitfeld, P. P. et al.: Does bladder preservation (as a surgical principle) lead to retaining bladder function in bladder/prostate rhabdomyosarcoma? Results from intergroup rhabdomyosarcoma study iv. J Urol, 171: 2396, 2004
37. Merguerian, P. A., Agarwal, S., Greenberg, M. et al.: Outcome analysis of rhabdomyosarcoma of the lower urinary tract. J Urol, 160: 1191, 1998
38. Chui, C. H., Spunt, S. L., Liu, T. et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg, 37: 1424, 2002
39. Heyn, R., Newton, W. A., Raney, R. B. et al.: Preservation of the bladder in patients with rhabdomyosarcoma. J Clin Oncol, 15: 69, 1997
40. Leuschner, I., Harms, D., Mattke, A. et al.: Rhabdomyosarcoma of the urinary bladder and vagina: a clinicopathologic study with emphasis on recurrent disease: a report from the Kiel Pediatric Tumor Registry and the German CWS Study. Am J Surg Pathol, 25: 856, 2001
41. Stevens, M. C., Rey, A., Bouvet, N. et al.: Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology--SIOP Malignant Mesenchymal Tumor 89. J Clin Oncol, 23: 2618, 2005
42. Yeung, C. K., Ward, H. C., Ransley, P. G. et al.: Bladder and kidney function after cure of pelvic rhabdomyosarcoma in childhood. Br J Cancer, 70: 1000, 1994
43. Ozkan, K. U., Bauer, S. B., Khoshbin, S. et al.: Neurogenic bladder dysfunction after sacrococcygeal teratoma resection. J Urol, 175: 292, 2006
44. Mosiello, G., Gatti, C., De Gennaro, M. et al.: Neurovesical dysfunction in children after treating pelvic neoplasms. BJU Int, 92: 289, 2003
45. Cruccetti, A., Kiely, E. M., Spitz, L. et al.: Pelvic neuroblastoma: low mortality and high morbidity. J Pediatr Surg, 35: 724, 2000
46. Ritchey, M., Ferrer, F., Shearer, P. et al.: Late effects on the urinary bladder in patients treated for cancer in childhood: a report from the Children's Oncology Group. Pediatr Blood Cancer, 52: 439, 2009
47. Heyn, R., Raney, R. B., Jr., Hays, D. M. et al.: Late effects of therapy in patients with paratesticular rhabdomyosarcoma. Intergroup Rhabdomyosarcoma Study Committee. J Clin Oncol, 10: 614, 1992
48. Hale, G. A., Marina, N. M., Jones-Wallace, D. et al.: Late effects of treatment for germ cell tumors during childhood and adolescence. J Pediatr Hematol Oncol, 21: 115, 1999
49. Dorr, W., Beck-Bornholdt, H. P.: Radiation-induced impairment of urinary bladder function in mice: fine structure of the acute response and consequences on late effects. Radiat Res, 151: 461, 1999
50. Tefft, M., Lattin, P. B., Jereb, B. et al.: Acute and late effects on normal tissues following combined chemo- and radiotherapy for childhood rhabdomyosarcoma and Ewing's sarcoma. Cancer, 37: 1201, 1976
51. Mangar, S. A., Foo, K., Norman, A. et al.: Evaluating the effect of reducing the high-dose volume on the toxicity of radiotherapy in the treatment of bladder cancer. Clin Oncol (R Coll Radiol), 18: 466, 2006
52. Hays, D. M., Raney, R. B., Wharam, M. D. et al.: Children with vesical rhabdomyosarcoma (RMS) treated by partial cystectomy with neoadjuvant or adjuvant chemotherapy, with or without radiotherapy. A report from the Intergroup Rhabdomyosarcoma Study (IRS) Committee. J Pediatr Hematol Oncol, 17: 46, 1995
53. Jerkins, G. R., Noe, H. N., Hill, D.: Treatment of complications of cyclophosphamide cystitis. J Urol, 139: 923, 1988
54. Filipas, D., Fisch, M., Stein, R. et al.: Rhabdomyosarcoma of the bladder, prostate or vagina: the role of surgery. BJU Int, 93: 125, 2004
55. Duel, B. P., Hendren, W. H., Bauer, S. B. et al.: Reconstructive options in genitourinary rhabdomyosarcoma. J Urol, 156: 1798, 1996
56. van Hemelrijck, M., Thorstenson, A., Smith, P. et al.: Risk of in-hospital complications after radical cystectomy for urinary bladder carcinoma: population-based follow-up study of 7608 patients. BJU Int, 112: 1113, 2013
57. Castagnetti, M., Angelini, L., Alaggio, R. et al.: Oncological outcome and urinary function after radical cystectomy for rhabdomyosarcoma in children: role of the orthotopic ileal neo-bladder based on a 15-year experience at a single centre. J Urol, In press, 2013
58. Austen, M., Kalble, T.: Secondary malignancies in different forms of urinary diversion using isolated gut. J Urol, 172: 831, 2004
59. Hassan, E., Creatsas, G., Michalas, S.: Genital tumors during childhood and adolescence. A clinical and pathological study of 71 cases. Clin Exp Obstet Gynecol, 26: 20, 1999
60. Hellman, K., Silfversward, C., Nilsson, B. et al.: Primary carcinoma of the vagina: factors influencing the age at diagnosis. The Radiumhemmet series 1956-96. Int J Gynecol Cancer, 14: 491, 2004
61. Arndt, C. A., Donaldson, S. S., Anderson, J. R. et al.: What constitutes optimal therapy for patients with rhabdomyosarcoma of the female genital tract? Cancer, 91: 2454, 2001
62. Fernandez-Pineda, I., Spunt, S. L., Parida, L. et al.: Vaginal tumors in childhood: the experience of St. Jude Children's Research Hospital. J Pediatr Surg, 46: 2071, 2011
63. Kirsch, C. H., Goodman, M., Esiashvili, N.: Outcome of Female Pediatric Patients Diagnosed With Genital Tract Rhabdomyosarcoma Based on Analysis of Cases Registered in SEER Database Between 1973 and 2006. Am J Clin Oncol, 37: 47, 2014
64. Copeland, L. J., Sneige, N., Stringer, C. A. et al.: Alveolar rhabdomyosarcoma of the female genitalia. Cancer, 56: 849, 1985
65. Qualman, S. J., Coffin, C. M., Newton, W. A. et al.: Intergroup Rhabdomyosarcoma Study: update for pathologists. Pediatr Dev Pathol, 1: 550, 1998
66. Newton, W. A., Jr., Gehan, E. A., Webber, B. L. et al.: Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer, 76: 1073, 1995
67. Scarpato, K. R., Ferrer, F. A.: Genitourinary rhabdomyosarcoma in children. In: AUA Update Series, vol. 32 (9), 2013
68. Grimsby, G. M., Ritchey, M. L.: Pediatric urologic oncology. Pediatr Clin North Am, 59: 947, 2012
69. Youngstrom, E. A., Bartkowski, D. P.: Vulvar embryonal rhabdomyosarcoma: a case report. J Pediatr Urol, 9: e144, 2013
70. Hays, D. M., Shimada, H., Raney, R. B., Jr. et al.: Sarcomas of the vagina and uterus: the Intergroup Rhabdomyosarcoma Study. J Pediatr Surg, 20: 718, 1985
71. Andrassy, R. J., Hays, D. M., Raney, R. B. et al.: Conservative surgical management of vaginal and vulvar pediatric rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma Study III. J Pediatr Surg, 30: 1034, 1995
72. Martelli, H., Oberlin, O., Rey, A. et al.: Conservative treatment for girls with nonmetastatic rhabdomyosarcoma of the genital tract: A report from the Study Committee of the International Society of Pediatric Oncology. J Clin Oncol, 17: 2117, 1999
73. Rodeberg, D. A., Paidas, C. N., Lobe, T. L. et al.: Surgical Principles for Children/Adolescents With Newly Diagnosed Rhabdomyosarcoma: A Report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. Sarcoma, 6: 111, 2002
74. Breneman, J., Meza, J., Donaldson, S. S. et al.: Local control with reduced-dose radiotherapy for low-risk rhabdomyosarcoma: a report from the Children's Oncology Group D9602 study. Int J Radiat Oncol Biol Phys, 83: 720, 2012
75. Walterhouse, D. O., Meza, J. L., Breneman, J. C. et al.: Local control and outcome in children with localized vaginal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma committee of the Children's Oncology Group. Pediatr Blood Cancer, 57: 76, 2011
76. Puri, D. R., Wexler, L. H., Meyers, P. A. et al.: The challenging role of radiation therapy for very young children with rhabdomyosarcoma. Int J Radiat Oncol Biol Phys, 65: 1177, 2006
77. Raney, R. B., Jr., Gehan, E. A., Hays, D. M. et al.: Primary chemotherapy with or without radiation therapy and/or surgery for children with localized sarcoma of the bladder, prostate, vagina, uterus, and cervix. A comparison of the results in Intergroup Rhabdomyosarcoma Studies I and II. Cancer, 66: 2072, 1990
78. Magne, N., Haie-Meder, C.: Brachytherapy for genital-tract rhabdomyosarcomas in girls: technical aspects, reports, and perspectives. Lancet Oncol, 8: 725, 2007
79. Magne, N., Oberlin, O., Martelli, H. et al.: Vulval and vaginal rhabdomyosarcoma in children: update and reappraisal of Institut Gustave Roussy brachytherapy experience. Int J Radiat Oncol Biol Phys, 72: 878, 2008
80. Rodary, C., Gehan, E. A., Flamant, F. et al.: Prognostic factors in 951 nonmetastatic rhabdomyosarcoma in children: a report from the International Rhabdomyosarcoma Workshop. Med Pediatr Oncol, 19: 89, 1991
81. Vasquez, R., Collini, P., Meazza, C. et al.: Late relapse of embryonal rhabdomyosarcoma, botryoid variant, of the vagina. Pediatr Blood Cancer, 51: 140, 2008
82. Spunt, S. L., Sweeney, T. A., Hudson, M. M. et al.: Late effects of pelvic rhabdomyosarcoma and its treatment in female survivors. J Clin Oncol, 23: 7143, 2005
83. Ariza, M., Rafaee, T., Adeeb, N. et al.: A successful pregnancy outcome in treated vulval rhabdomyosarcoma. Med J Malaysia, 54: 371, 1999
84. Mansky, P., Arai, A., Stratton, P. et al.: Treatment late effects in long-term survivors of pediatric sarcoma. Pediatr Blood Cancer, 48: 192, 2007
85. Kenney, L. B., Laufer, M. R., Grant, F. D. et al.: High risk of infertility and long term gonadal damage in males treated with high dose cyclophosphamide for sarcoma during childhood. Cancer, 91: 613, 2001
86. Ridola, V., Fawaz, O., Aubier, F. et al.: Testicular function of survivors of childhood cancer: a comparative study between ifosfamide- and cyclophosphamide-based regimens. Eur J Cancer, 45: 814, 2009
87. Oberlin, O., Fawaz, O., Rey, A. et al.: Long-term evaluation of Ifosfamide-related nephrotoxicity in children. J Clin Oncol, 27: 5350, 2009
88. Hishiki, T., Saito, T., Mitsunaga, T. et al.: Optimal surgical treatment and urological outcomes in boys with pelvic and urogenital rhabdomyosarcomas and soft tissue sarcomas. Pediatr Surg Int, 29: 1077, 2013
89. Raney, B., Anderson, J., Arndt, C. et al.: Primary renal sarcomas in the Intergroup Rhabdomyosarcoma Study Group (IRSG) experience, 1972-2005: A report from the Children's Oncology Group. Pediatr Blood Cancer, 51: 339, 2008
90. Fanous, R. N., Mayer, E. K., Vale, J. et al.: Primary renal embryonal rhabdomyosarcoma in adults: a case report and review of the literature. Case Rep Oncol Med, 2012: 460749, 2012
91. Carpentieri, D. F., Nichols, K., Chou, P. M. et al.: The expression of WT1 in the differentiation of rhabdomyosarcoma from other pediatric small round blue cell tumors. Mod Pathol, 15: 1080, 2002
92. Morotti, R. A., Nicol, K. K., Parham, D. M. et al.: An immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the Children's Oncology Group experience. Am J Surg Pathol, 30: 962, 2006
93. Walther, A., Cost, N. G., Garrison, A. P. et al.: Renal rhabdomyosarcoma in a pancake kidney. Urology, 82: 458, 2013
94. Blakely, M. L., Andrassy, R. J., Raney, R. B. et al.: Prognostic factors and surgical treatment guidelines for children with rhabdomyosarcoma of the perineum or anus: a report of Intergroup Rhabdomyosarcoma Studies I through IV, 1972 through 1997. J Pediatr Surg, 38: 347, 2003
95. Pham, T. H., Iqbal, C. W., Zarroug, A. E. et al.: Retroperitoneal sarcomas in children: outcomes from an institution. J Pediatr Surg, 42: 829, 2007









