









Rabdomiosarcoma genitourinario
Nicholas G. Cost, MD1, Jonathan C. Routh, MD, MPH2 y Kathleen Kieran, MD3
1División de Urología, Facultad de Medicina de la Universidad de Colorado, Aurora, CO
2División de Cirugía Urológica, Duke University Medical Center, Durham, NC
3Departamento de Urología, Hospitales y Clínicas de la Universidad de Iowa, Iowa City, IA
Traducido y editado desde el original al español | Enlace a la versión en inglés
Dra Luciana Lerendegui, Dr Juan Manuel Modes
Hospital Italiano, Buenos Aires, Argentina
Introducción
Elrabdomiosarcoma (RMS) es el sarcoma de partes blandas más frecuente en bebés y niños, con aproximadamente 350 casos nuevos en los Estados Unidos cada año. Es el tercer tumor sólido más común en los niños después del neuroblastoma y el tumor de Wilms, representando el 10-15% de todos los tumores pediátricos sólidos.1 Por definición, el RMS es un sarcoma con diferenciación a músculo esquelético pero que puede ocurrir en sitios donde no se encuentra dicho tejido, lo que sugiere que es un tumor mesenquimático primitivo condiferenciación a músculo esquelético.2 La incidencia de RMS sigue un patrón de edad bimodal. La primera ventana de mayor incidencia, que representa más del 50% de los casos, ocurre en la primera década. Este período alcanza su punto máximo en una mediana de 2 años de edad, y consiste principalmente en RMS embrionario (E-RMS) o botrioide (B-RMS). El segundo pico ocurre durante la adolescencia y más comúnmente consiste en RMS alveolar (A-RMS).1 Además de variar su edad en la presentación, las variantes histopatológicas de RMS (embrionario, botrioide y alveolar) también se asocian con diferentes patrones de presentación primaria y resultados posteriores. El E-RMS tiende a ocurrir en la cabeza / cuello y en el sistema genitourinario (GU) y la mayoría de los pacientes sobreviven más de 5 años. El A-RMS se ve a menudo en las extremidades y tiende a metastatizarse temprano con una peor supervivencia que la E-RMS.3
Desde su descubrimiento en 1854, el estudio de las características clínicas y patológicas del RMS ha llevado progresivamente al desarrollo de criterios de diagnóstico uniformes y sistemas de estadificación.4 La perspectiva pronóstica para RMS ha mejorado significativamente con el tiempo, desde una supervivencia estimada del 25% en 1970 hasta más del 70% en la actualidad.1 Este éxito se debe a una terapia multimodal efectiva, adaptada al riesgo y una mejor atención de apoyo. La información emergente sobre la biología del tumor ha brindado nuevos conocimientos sobre la patogénesis de este tumor y ha permitido avances en el manejo basado en el riesgo.
RMS genitourinario
Entre el 15 y el 20 por ciento de todos los casos de RMS surgen del sistema GU.5 Los sitios GU más comunes son próstata, vejiga y paratestisticular. Los RMS vaginal, cervical y uterino son sitios relativamente inusuales. Al igual que con RMS en general, las tasas de supervivencia varían según el sitio dentro del subgrupo de GU-RMS. Específicamente, el RMS vaginal y paratesticular tienen un mejor pronóstico que el RMS de vejiga / próstata.
Epidemiología / Factores de riesgo
Casi el 75% de los casos de GU-RMS se diagnostican antes de los 5 años, con un predominio masculino de 3,3:1.6 La edad de diagnóstico afecta el pronóstico independientemente de la histología del tumor, y los pacientes <1 año y> 10 años tienen una peor tasa de sobrevida sin complicaciones en comparación con aquellos entre 1 y 9 años (53% y 51%, vs 71%, respectivamente).7 Existen factores de riesgo establecidos para desarrollar RMS y dada la corta edad de los afectados; estos son en su mayoría congénitos en contraposición a los factores de riesgo adquiridos. El síndrome de Costello, el síndrome de nevus de células basales de Gorlin, el síndrome de Rubinstein-Taybi, el síndrome de trisomía 21, el síndrome de Beckwith-Wiedemann y el síndrome de alcoholismo fetal se han asociado con RMS.8 Dos afecciones genéticas requieren una mención particular: (1) la neurofibromatosis es el síndrome más común en el que se ha descrito el RMS. De los pacientes incluidos en el Estudio IV de Intergrupos de Rabdomiosarcoma (IRS IV), la incidencia de neurofibromatosis fue del 0,5%.9 (2) El síndrome de Li-Fraumeni es una mutación de la línea germinal del gen supresor de tumores TP53 y se transmite de manera autosómica dominante. Se ha encontrado un aumento de la incidencia de RMS en asociación con el síndrome de Li-Fraumeni, al igual que otros tumores sólidos en la infancia, incluido el carcinoma adrenocortical.10 Los factores ambientales asociados con el RMS incluyen la exposición a la radiación y la quimioterapia previa con agentes alquilantes. El aumento del riesgo de neoplasias malignas secundarias en pacientes con neurofibromatosis y síndromes de Li-Fraumeni después del tratamiento con agentes alquilantes y la radioterapia para RMS establece un vínculo entre la genética y el medio ambiente. Por lo tanto, es deseable limitar el uso de agentes alquilantes y radioterapia en estas poblaciones particulares.11
Biología del tumor / Genética
La histología de RMS consiste de pequeñas células azules redondas, con números variables de células fusiformes que se parecen al músculo esquelético fetal. El Intergroup Rhabdomyosarcoma Study Group (IRSG) ha desarrollado una clasificación que reconoce tres grupos histológicos principales que tienen importancia pronóstica.12 (1) La histología embrionaria, que es favorable, representa el 90% del RMS genitourinario. (2) La histología botrioide, que se ve a menudo en la RMS vesical y vaginal, es esencialmente una variante de la E-RMS. (3) La histología alveolar representa el 10% restante de GU-RMS y se asocia con los peores resultados.3 El E-RMS puede presentarse en una forma sólida que surge en grupos musculares, como el tronco y las extremidades, o como sarcoma botrioides, una variedad polipoide que se presenta en órganos huecos o cavidades corporales, como la vejiga o la vagina. Hay una célula fusiforme, o variante leiomiomatosa, que se ve con frecuencia en la región paratesticular. Las variantes botrioides y de células fusiformes de E-RMS se asocian con una excelente supervivencia.
El diagnóstico patológico de RMS va más allá de la clasificación histológica básica y se complementa con técnicas como la inmunohistoquímica (IHC) y la citogenética. Específicamente, la tinción IHC ha sido útil en el diagnóstico y pronóstico del A-RMS. Este último, demuestra altos niveles de tinción con miogenina, mientras que el E-RMS es negativo o solo tiene tinción de bajo nivel.13 Posteriormente, se ha demostrado que la tinción con miogenina difusa predice la disminución de la sobrevida independientemente del estadio del tumor, el sitio o la histología.14 Además, se ha observado que el E-RMS y el A-RMS tienen distintas anomalías citogenéticas. El A-RMS se asocia con una translocación entre los cromosomas 1 o 2 y 13, lo que resulta en la formación de una proteína quimérica. PAX3, una proteína de unión a ADN en el cromosoma 2, o PAX7, una proteína de unión a ADN en el cromosoma 1, se fusiona con el gen FKHR en el cromosoma 13.1516 La expresión de la translocación t(2; 13), PAX3-FKHR es un factor pronóstico adverso para los niños que presentan A-RMS metastásico. Además, los pacientes con la translocaciónt(1; 13), que presentan el producto de fusión PAX7-FKHR, a menudo son más jóvenes y tienen un mejor pronóstico que sus contrapartes con la t (2; 13), anomalía de la fusión PAX3-FKHR .1617 Esto ha llevado a algunos a proponer una estratificación de riesgo modificada que incluye este estado de translocación.18 Sin embargo, el análisis de una gran serie de casos no logra mostrar una asociación absoluta de las translocaciones PAX-FHKR con A-RMS. Al menos el 25% de estos tumores poseen una histología alveolar clásica pero carecen de una translocación. En contraste, el E-RMS no demuestra translocaciones cromosómicas recurrentes. En cambio, muestran una mayor inestabilidad genómica y desequilibrios alélicos recurrentes, como la pérdida de heterocigosidad (LOH) en el cromosoma 11p15.5. Esto ocurre en una ubicación diferente a la del Gen WT2 implicado en el desarrollo de algunos tumores de Wilms.19 Esta región es donde se ubica el gen IGF-2. La sobreexpresión de IGF-2 se ha documentado tanto en el A-RMS como en el E-RMS. El IGF-2 es un factor de crecimiento que puede estimular el crecimiento de las células tumorales del RMS, y los anticuerpos dirigidos contra el IGF-2 pueden inhibir el crecimiento del tumor.20 La amplificación del factor de transcripción N- MYC se ha observado en el RMS y la sobreexpresión de N- MYC se asoció con un resultado adverso en A-RMS.21 En resumen, utilizando estas técnicas avanzadas y complementarias a la histopatología estándar, es posible la clasificación basada en la expresión génica utilizando tan solo cinco genes con una tasa de error estimada de menos del 5%.19 Con ese fin, la próxima ronda de estudios del Children's Oncology Group (COG) para RMS probablemente usará el estado de fusión por translocación como factor de riesgo biológico sustituto en lugar de histología embrionaria o alveolar, es decir, la histología alveolar sin fusión por translocación se manejará como una Tumor E-RMS.
Estadificación/agrupación/clasificación de riesgo para GU-RMS
El enfoque para clasificar a los pacientes con RMS puede parecer inicialmente complicado. Sin embargo, utilizando factores importantes que predicen pronóstico, estos sistemas intentan estadificar lógicamente a los pacientes antes de la cirugía, agruparlos basándose en la integridad de la resección y estratificarlos por riesgo según la histología, el estadio y el grupo (Tablas I , II y III). El sistema Tumor-Node-Metastasis (TNM) segrega a los pacientes por sitios favorables y desfavorables y ahora se utiliza en una estadificación IRSG integrada. Los sitios GU favorables incluyen paratesticular y Vaginal / Cervical / Uterino. Los sitios GU desfavorables son la vejiga y la próstata. La presencia de metástasis a distancia en el momento del diagnóstico, ganglios linfáticos regionales afectados (LN) y tumores primarios grandes (> 5 cm) en sitios desfavorables son signos pronósticos relativamente desfavorables.22 Un elemento importante a tener en cuenta al evaluar a pacientes individuales es que la agrupación posquirúrgica depende en gran medida de la integridad de la escisión quirúrgica. A medida que el tratamiento del GU-RMS ha evolucionado, más pacientes con RMS de vejiga / próstata se someten a una biopsia sólo en el procedimiento quirúrgico inicial, lo que deja una enfermedad residual grave. Esto resulta en el desplazamiento de más pacientes del grupo I al grupo III. Por lo tanto, los tumores teóricamente equivalentes podrían terminar en diferentes grupos, dependiendo de la agresividad de la resección quirúrgica inicial. Los estudios IRSG y COG RMS actuales agrupan a los pacientes en grupos de riesgo bajo, intermedio y alto. Esta clasificación de riesgo combina la agrupación clínica y el sistema TNM con la adición de histología. El sistema intenta clasificar a los pacientes según las diversas variables pronósticas conocidas para predecir los resultados. Un cambio reciente en este sistema ocurrió con el estudio ARG0431 del COG que incluyó todas las edades de E-RMS en Etapa IV en el grupo de alto riesgo. Anteriormente, los pacientes con E-RMS en estadio IV menores de 10 años de edad se consideraban de riesgo intermedio.
Paradigma de tratamiento general
El tratamiento del GU-RMS ha evolucionado desde la escisión quirúrgica radical hacia métodos de preservación de órganos cuando es posible. Una vez que se demostró que la mayoría de los pacientes sobreviven a la enfermedad, los investigadores exploraron el uso de la quimioterapia primaria y la radioterapia para evitar la exenteración quirúrgica generalmente empleada para el GU-RMS. Los estudios más recientes se han centrado en una mayor morbilidad y reducción de la terapia. La actual ronda de estudios del COG ha concluido recientemente y los resultados se anticiparán en los próximos años. Para los pacientes de bajo riesgo con RMS embrionario / botrioide / de células fusiformes, se inauguró en 2004 el ARST0331. Este estudio evaluó la reducción de la duración de la quimioterapia para los tumores en estadio favorable de estadio bajo. El estudio para pacientes de riesgo intermedio, ARST0531, evaluó el papel de la radiación anterior.
Si bien la tendencia ha sido generalmente hacia la reducción de la terapia, los estudios han indicado ocasionalmente que la terapia debe intensificarse. Por ejemplo, si bien parece que a los niños menores de 10 años con RMS paratesticular se les puede evaluar el retroperitoneo en forma segura con imágenes de corte transversal, aquellos de ≥ 10 años de edad tienen una estadificación insuficiente con imágenes solamente. Por lo tanto, se requiere una disección ipsilateral de ganglios linfáticos retroperitoneales (RPLND) en ese grupo de edad.2324 Del mismo modo, la intensificación terapéutica está bajo investigación para aquellos que experimentaron resultados deficientes en estudios anteriores. ARST0431, el estudio para pacientes de alto riesgo, investigó regímenes de quimioterapia más intensivos en un intento por mejorar la supervivencia en esta población.
RMS paratesticular
Antecedentes
El RMS paratesticular (PT-RMS, por sus siglas en inglés) representa entre el 5 y el 10% de todos los diagnósticos nuevos de RMS y ocurre en una distribución de edades bimodal, con un diagnóstico de aproximadamente 1/3 entre las edades de 1 a 5 años y otro 1/3 diagnosticado después de los 10 años de edad.2526 PT-RMS surge en el músculo de la porción distal del cordón espermático y puede invadir el testículo o los tejidos circundantes. La presentación es a menudo una hinchazón o masa escrotal indolora que es distinta del testículo. Debido a esta ubicación superficial y la facilidad con la que se puede examinar y observar, el PT-RMS generalmente se detecta y diagnostica antes. Por ejemplo, entre el 60 y el 80% de los tumores PT se encuentran en la etapa 1, en comparación con el 10 al 15% de todas las ubicaciones primarias de RMS.26 Más del 90% de los casos de PT-RMS tienen histología embrionaria y un buen pronóstico. Sin embargo, independientemente de la histología, se considera una ubicación favorable porque incluso los pacientes con PT-RMS alveolar tienen un mejor pronóstico que otras primarias de RMS alveolar.27
Presentación clínica y manejo inicial.
El paciente o su familia suelen observar el PT-RMS como una masa escrotal indolora que puede o no ser distinta del testículo. El enfoque clínico de esta situación implica una historia clínica completa y un examen físico seguido de marcadores tumorales séricos para evaluar una neoplasia testicular primaria (AFP, bHCG, LDH), así como una ecografía escrotal para tratar de distinguir entre una ubicación primaria testicular o paratesticular. Es de destacar que el examen físico debe tener en cuenta específicamente cualquier invasión aparente de la pared escrotal, así como cualquier linfadenopatía inguinal o cervical. La estadificación consiste en electrolitos séricos, hemograma completo, pruebas de función hepática y aspirados / biopsias de médula ósea bilateral. La estadificación adicional incluye imágenes de corte transversal del tórax, y abdomen. (Figura 1).
Imágenes de TC pélvica axial de tumor primario en un niño con RMS paratesticular
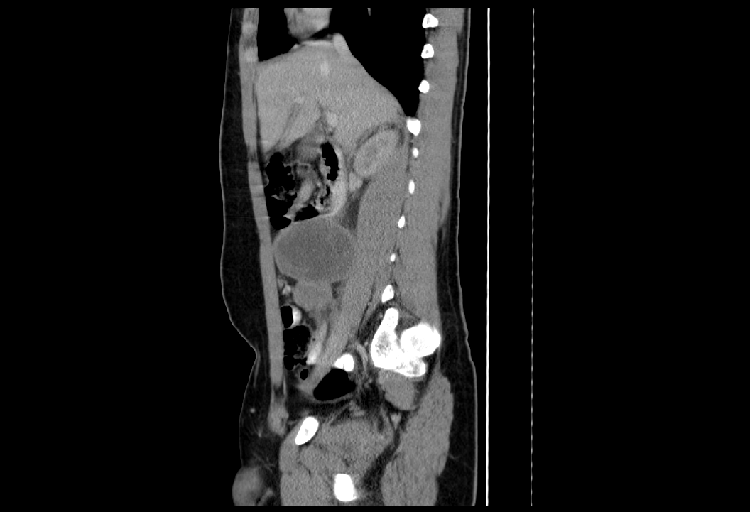
Figura 1. Imágenes de TC abdominal sagital de linfadenopatía masiva en un niño con RMS paratesticular
Figura 1. Imágenes de TC abdominal sagital de linfadenopatía masiva en un niño con RMS paratesticular
Las exploraciones óseas han sido tradicionalmente parte de la estadificación en el momento del diagnóstico según los protocolos del grupo de estudio cooperativo. Sin embargo, una investigación reciente sugiere que las exploraciones con FDG-PET pueden ser más beneficiosas en la estadificación y para futuras comparaciones durante y después del tratamiento para evaluar la efectividad terapéutica (Figura 2).2829
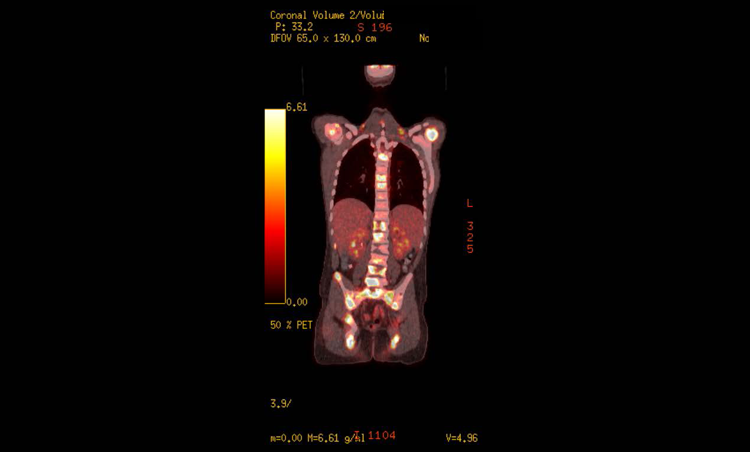 Figura 2 Imágenes PET de metástasis óseas y blandas generalizadas en un niño con RMS paratesticular
Figura 2 Imágenes PET de metástasis óseas y blandas generalizadas en un niño con RMS paratesticular
Orquiectomía
Independientemente de la presencia o ausencia de enfermedad metastásica en las imágenes por etapas, el siguiente paso en el tratamiento es la escisión de la lesión primaria, que siempre debe abordarse mediante una orquiectomía inguinal radical. No hay ningún papel para la exploración escrotal y dicha cirugía exigiría una escisión secundaria de esta cicatriz escrotal, así como una incisión inguinal para resecar el cordón espermático inguinal restante. La integridad de esta resección tendrá un impacto en el manejo posterior, incluida la dosificación de radiación.
Terapia adyuvante
Disección ganglionar retroperitoneal (RPLND)
La cirugía adicional en este punto depende de la edad del paciente y de las imágenes de la estadificación inicial. Sin embargo, estudios previos que investigaron la estadificación ipsilateral de RPLNDdemostraron que niños de 10 años de edad o más tenían un mayor riesgo de ser subestimados solo por imágenes que los menores de 10 años.232430 Por lo tanto, la recomendación actual es realizar RPLND ipsilateral a todos los niños de 10 años de edad o mayores. La RPLND puede realizarse de forma tradicional, abierta o mediante técnicas mínimamente invasivas por parte de cirujanos experimentados con este enfoque.3132 Además, los niños de cualquier edad que presenten metástasis retroperitoneales deben recibir RPLND. La excepción a este plan de tratamiento es la presencia de afectación ganglionar voluminosa (más de 2 masas mayores de 2 cm). En el contexto de dicha enfermedad avanzada en la imagenología, la biopsia puede confirmar la metástasis para permitir la estadificación adecuada y el inicio rápido de la terapia. La realización de la RPLND después de la quimioterapia de inducción está indicada si los primeros ciclos de quimioterapia han permitido una reducción suficiente para que una RPLND pueda eliminar con seguridad toda la enfermedad grave. Si este RMS metastásico se reseca del retroperitoneo, entonces se administra radioterapia ajustada a la dosis. Si estas metástasis retroperitoneales no se resecan, entonces se administra radiación a 5040cGy a las masas residuales y se puede considerar nuevamente RPLND para resecar el residuo al finalizar toda la quimioterapia y radiación en caso de ser tales masas aún radiográficamente visibles.
Quimioterapia y Radiación
Actualmente hay estudios en curso para evaluar la reducción de la quimioterapia en pacientes con PT-RMS embrionario porque generalmente representan una población de bajo riesgo. Del mismo modo, hay estudios que investigan la intensificación de la dosis para el PT-RMS alveolar. Sin embargo, estas recomendaciones en este capítulo siguen los estándares de atención más actuales. Independientemente de la histología, los pacientes con PT-RMS que son N0 o N1, por imágenes si tienen menos de 10 años de edad o por RPLND si tienen 10 años de edad o más, y no tienen evidencia de metástasis a distancia se tratan con 45 semanas de vincristina, actinomicina D y ciclofosfamida (VAC). Debe mencionarse que los pacientes con RMS suelen recibir a menudo ciclofosfamida o ifosfamida y corren el riesgo de cistitis hemorrágica y otros efectos tardíos sobre la vejiga y, por lo tanto, se manejan con hiperhidratación y MESNA en el momento de la administración de quimioterapia.
Los pacientes del grupo I en estadio 1 (N0) no requieren radioterapia. Los pacientes con PT-RMS del grupo II localizado representan a aquellos que tuvieron un derrame local en el sitio paratesticular. Estos pacientes requieren radioterapia en este sitio, que tradicionalmente corresponde a 4140cGy. Sin embargo, si la enfermedad permanece en el sitio primario, se debe considerar la cirugía de revisión, que puede incluir hemi-escrotectomía, para obtener la resección de la enfermedad residual. Esto permitiría una reducción de la dosis a 3600 cGy en el sitio del PT si se obtiene una resección completa. Además, al igual que cualquier paciente con PT-RMS, si los pacientes del grupo II localizado tienen evidencia de enfermedad ganglionar retroperitoneal metastásica, también necesitarán radioterapia retroperitoneal. La dosis de radiación al retroperitoneo depende del estado de estos nodos. Si los ganglios se extirpan por completo en RPLND y son positivos (grupo II retroperitoneal), esta dosis es 4140 cGy. Sin embargo, si hay enfermedad residual grave en el retroperitoneo (Grupo III), ya sea por escisión incompleta o solo por biopsia, esta dosis se incrementa a 5040 cGy.
Para la enfermedad ampliamente metastásica (es decir, no solo en el retroperitoneo), el paradigma del tratamiento es bastante diferente. Estos pacientes se consideran de alto riesgo independientemente de su histología. Anteriormente, los pacientes en estadio 4 menores de 10 años con E-RMS se consideraban de riesgo intermedio. En los estudios actuales, se incluye a estos pacientes con todas las histologías y la enfermedad del grupo IV en estadio 4 para clasificarlos como de alto riesgo. Estos pacientes deben someterse a una orquiectomía inguinal radical para el diagnóstico, seguido de enrolamiento en el estudio. Todos estos pacientes ahora reciben 51 semanas de quimioterapia intensiva con VAC más Irinotecan, Ifosfamide, Etoposide y Doxorubicin. El plan de radioterapia depende de la presencia y el volumen de la enfermedad metastásica en el retroperitoneo y la integridad de la resección en el momento de la orquiectomía. Como se mencionó anteriormente, la estadificación de la RPLND tiene un papel en la evaluación de la diseminación retroperitoneal, excepto en aquellos pacientes con enfermedad voluminosa (más de 2 masas mayores de 2 cm). En el contexto de una enfermedad más avanzada en la imagenología, la biopsia de masa retroperitoneal inicial puede confirmar la metástasis y permitir una evaluación rápida del riesgo y el inicio de la terapia. La RPLND finalizada después de la quimioterapia de inducción está indicada si las primeras 20 semanas de quimioterapia permiten una reducción suficiente de la enfermedad tal que la RPLND podría eliminar toda la enfermedad grave. Si este RMS metastásico se reseca, se administra radioterapia. Sin embargo, si estas metástasis retroperitoneales no se resecan, la radiación a 5040cGy se administra primero a estos nodos residuales. La RPLND para la resección de este residuo puede ser considerada nuevamente al finalizar las 51 semanas de quimioterapia si aún esas masas son radiográficamente evidentes. Todos los demás sitios no metódicos de metástasis reciben 5040 cGy de radiación.
En cuanto al control local en el contexto de la enfermedad en estadio IV, esto es similar a aquellos con enfermedad en estadio I. Es decir, si se produce un derrame en el momento de la orquiectomía y persisten enfermedades graves, una segunda operación que puede incluir una hemiscrotectomía para resecar esta enfermedad puede permitir una reducción de la radioterapia local requerida. Para los pacientes con PT-RMS y enfermedad completamente resecada en el sitio primario de una orquiectomía inguinal radical con márgenes negativos, no se administra radiación. Para aquellos que tienen enfermedad microscópica paratesticular o enfermedad residual gruesa, se administra dirigida a los campos inguinal y escrotal.
Pronóstico
El pronóstico para los pacientes con PT-RMS es generalmente excelente en el contexto de una enfermedad localizada (Etapa 1 Grupo I-III).26 El mejor pronóstico se observa en aquellos con enfermedad de los Grupos I y II en estadio 1, donde la sobrevida global y la sobrevida libre de eventos son del 94 al 96% y del 91 al 95%, respectivamente. Las personas con enfermedad del Grupo III tienen un pronóstico ligeramente peor con sobrevida libre de eventos a 5 años y sobrevida global del 75% y 76%, respectivamente. Estos resultados parecen ser algo independientes de la histología alveolar o embrionaria.27 Los pacientes con PT-RMS alveolar y enfermedad localizada (Etapa 1 Grupo I-III) tuvieron una sobrevida libre de eventos y sobrevida global a 5 años de 78% y 89%, respectivamente.27 Sin embargo, queda por verse si estos pacientes con PT-RMS alveolar tienen más probabilidades de tener la translocación t (1; 13),fusión PAX7-FKHR, que puede no estar asociada con los resultados pobres más típicos de los pacientes mayores con A- RMS que generalmente tienen la anomalía de fusión t (2; 13), PAX3-FKHR . Independientemente de dicha evaluación de riesgo histopatológico, los pacientes con enfermedad metastásica en estadio IV tienen un peor pronóstico con una sobrevida global a 5 años, que se informa aproximadamente entre el 20 y el 25%.26
RMS de Vejiga y / o de próstata
Epidemiología
La vejiga y la próstata son los sitios más comunes de GU-RMS (VP-RMS), representando 5% del total de RMS. El RMS es la neoplasia de vejiga más común en niños menores de 10 años de edad; la edad media de los niños que lo presentanes de 5 años.30
Presentación clínica y evaluación inicial
El VP-RMS generalmente se presenta con hematuria, polaquiuria,y/o retención urinaria aguda.33 Como regla general, se debe considerar que un niño en edad preescolar con retención urinaria aguda tiene un alto riesgo de RMS y debe tratarse en consecuencia. En niñas con sarcoma botrioides intraluminal, el tumor puede ser visible a través de prolapso por el meato uretral (Figura 3).
Resultados del examen físico en dos niñas con sarcoma variante de botrioides de RMS
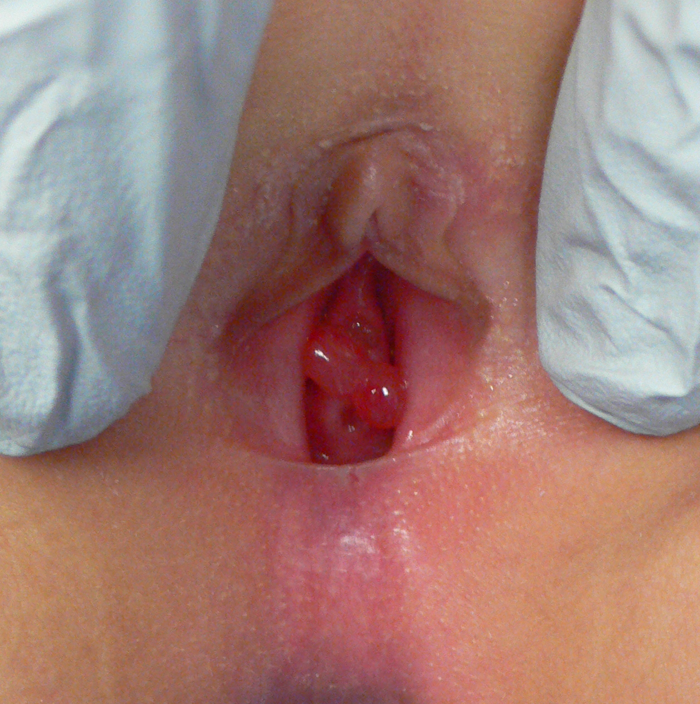
Figura 3
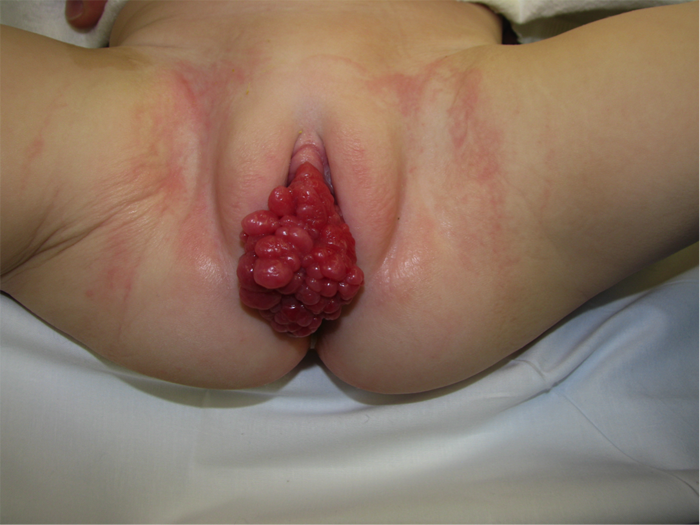
Figura 4
En el examen físico, a menudo está presente una masa abdominal inferior palpable, que representa la masa en sí misma, una vejiga distendida, o ambos. En particular, el RMS prostático es palpable en el examen rectal.
La diferenciación entre vejiga y próstata como el sitio de origen primario del cual surgió un RMS grande puede ser bastante desafiante. En general, las lesiones primarias prostáticas tienen más probabilidades de ser masas grandes y sólidas. Por el contrario, las lesiones primarias vesicales tienden a ocurrir a medida que se forma el sarcoma botrioides (Figura 4), estando estos tumores localizados más frecuentemente en el trígono o en el cuello de la vejiga.34
La evaluación inicial de un niño con sospecha de VP-RMS debe consistir en ultrasonido si la masa abdominal no es palpable. Si se puede palpar un tumor o una vejiga distendida, o si una ecografía inicial revela una masa sospechosa, se deben realizar imágenes de corte transversal con CT o MRI para determinar la extensión del tumor local ( Figura 5 ). Además, estas modalidades pueden evaluar el retroperitoneo en busca de evidencia de diseminación metastásica linfática o distante.
Tratamiento
Manejo quirúrgico inicial
Históricamente, el tratamiento habitual del VP-RMS fue la extirpación quirúrgica mediante cistoprostatectomía radical o, más comúnmente, exenteración pélvica.35 Comenzando con los estudios del IRSG en la década de 1970, sin embargo, la quimioterapia y la radiación comenzaron a suplantar a la cirugía como el pilar del tratamiento. De hecho, un objetivo importante del IRS-II (1978-1984) era preservar un tracto urinario funcional. Este enfoque se ha agudizado en los últimos años.36 Actualmente, los componentes principales del tratamiento quirúrgico son le biopsia inicial (idealmente por medios endoscópicos o percutáneos), seguida de quimioterapia y / o radioterapia, y por último, si es necesario y si es posible, cirugía.
El diagnóstico de VP-RMS obviamente requiere una revisión patológica de una muestra adecuada de tejido tumoral. Dependiendo del tamaño y la ubicación del tumor, esto debería lograrse por medios mínimamente invasivos, como la resección transuretral (el método preferido si es posible), la biopsia percutánea con aguja o la biopsia transrectal o transperineal guiada por ultrasonido. Ocasionalmente, estos métodos no son factibles, en cuyo caso puede ser necesaria una biopsia abierta a través de una incisión en la línea media baja o Pfannenstiel. En tales casos, se recomienda un intento de muestreo de ganglios linfáticos (como los nódulos ilíacos u obturadores) para mejorar la precisión de la estadificación.
En los casos en que el tumor se puede extirpar completamente con una morbilidad mínima, por ejemplo, en un paciente con un RMS pequeño en la cúpula de la vejiga, se puede considerar una cistectomía parcial con amplios márgenes locales.37 Una ventaja significativa de este enfoque es que, si se completa con márgenes negativos, el paciente puede ser capaz de retener una vejiga relativamente normal. En el IRS-IV, esta estrategia se aplicó con éxito en una serie de pacientes, y 13 de 17 niños se sometieron a una cistectomía parcial inicial sin evidencia de enfermedad en el seguimiento a largo plazo. La radioterapia fue necesaria en 10 de estos pacientes, sin embargo, con implicaciones para la función vesical postoperatoria.36
Si el tumor es lo suficientemente grande como para obstruir el uréter o el drenaje de la vejiga, se debe realizar una derivación urinaria temporal mediante un stent ureteral JJ, una sonda de nefrostomía percutánea y / o un catéter de Foley, según corresponda.33
Re-resección previa al tratamiento (RPT)
En casos raros, la re-resección previa al tratamiento, a menudo por cistectomía parcial, cistectomía o cistoprostatectomía, puede considerarse si es posible una escisión completa. Aunque la re-resección previa al tratamiento puede no ser necesaria en tumores no RMS, 38 este tratamiento no se emplea típicamente en VP-RMS. Dicho esto, los pacientes tratados con una resección no planificada o incompleta en la que RPT puede dejar al paciente libre de enfermedad, son potencialmente candidatos para este enfoque.
Cirugía de Second Look (SLO)
Después de la quimioterapia y / o la radioterapia, puede haber masas residuales. En estos casos, con frecuencia se necesita un procedimiento de revisión, que incluye, entre otros, la cistoscopia, la biopsia, la exploración, la cistectomía parcial, la cistoprostatectomía o la prostatectomía. En el IRS-IV, 53 de 88 pacientes con VP-RMS finalmente requirieron al menos una operación de second look.36 Sin embargo, evaluar la integridad de la respuesta terapéutica puede ser un desafío. Los rabdomioblastos maduros no se presentan con poca frecuencia en las biopsias posteriores al tratamiento, y se pueden confundir fácilmente con la enfermedad activa (en particular en la biopsia por congelación). Aunque hay una tasa de recurrencia generalmente baja de los rabdomioblastos maduros, se han informado 39 muertes después de este diagnóstico.40
Función vesical posterior al tratamiento
Como se indicó anteriormente, los pacientes con VP-RMS se manejan con cirugía intensiva, quimioterapia y / o radioterapia. La radioterapia, en particular, tiene un papel particularmente importante en los algoritmos de esta entidad. Tanto en el protocolo IRS-IV como en el protocolo SIOP MMT89, se observaron mayores tasas de recurrencia local en pacientes que no recibieron radiación.3641 Sin embargo, las tasas de sobrevida global fueron similares entre los que recibieron y no recibieron radiación.41 Sin embargo, la radioterapia puede tener un impacto sustancial en la función de la vejiga. En el IRS-IV, se informó que el 55% de los pacientes supervivientes sin eventos tenían la función de vejiga "preservada". Sin embargo, esta cifra se determinó basándose únicamente en el cuestionario del paciente, y sólo 1 niño se sometió a pruebas formales urodinámicas.36 En una serie más pequeña informada por Yeung et al, todos los pacientes que recibieron irradiación pélvica tenían perfiles urodinámicos notablemente anormales, reducían específicamente la capacidad funcional y las curvas de evacuación atípicas.42 Entre todos los niños con VP-RMS en el IRS-IV, solo el 40% no tuvo eventos con vejigas con funcionamiento normal, y como se señaló anteriormente, esto probablemente sea una sobreestimación dada la falta de evaluaciones formales urodinámicas en estos niños.36
Las anomalías en la función de la vejiga pueden surgir como consecuencia de una intervención quirúrgica (p. Ej., Daño a los nervios, cistectomía parcial) o tratamiento médico.434445 Las quejas típicas de presentación incluyen síntomas del tracto urinario inferior, incluida la enuresis. Ésta última puede ser más común en niños con antecedentes de RMS genitourinario que en sobrevivientes de otros cánceres infantiles.42 evaluación debe incluir no sólo una evaluación de la gravedad y molestia de los síntomas, sino también la integridad de los tractos superiores, ya que los hábitos de eliminación anormales (ya sea en la fase de almacenamiento o evacuación, o ambos) pueden colocar unidades renales ya expuestas a quimioterapia y / o radiación en riesgo.
La cistitis hemorrágica, que a menudo se desarrolla agudamente en pacientes que reciben quimioterapia con agentes alquilantes, también puede presentarse como un efecto tardío, con una presentación muy variable.46 La cistitis inducida por radiación y quimioterapia está correlacionada histológicamente con la hipoxia, isquemia y necrosis del tejido local. La ciclofosfamida y su metabolito acroleína se consideran los agentes etiológicos clásicos, pero otros agentes quimioterapéuticos (por ejemplo, dactinomicina y doxorubicina) pueden actuar de manera sinérgica para aumentar el riesgo de cistitis hemorrágica.474849 Además, los agentes que actúan para disminuir la toxicidad de los agentes quimioterapéuticos (por ejemplo, MESNA, N-acetilcisteína) no eliminan completamente el riesgo de cistitis hemorrágica. Los pacientes que reciben radiación pélvica parecen tener un mayor riesgo de cistitis hemorrágica, especialmente cuando las dosis superan los 3000 cGy para la exposición total de la vejiga y 6000 cGy para la exposición parcial de la vejiga.5051 Las dosis superiores a 4500 cGy se asocian con un desarrollo más constante de la cistitis por radiación.5152 Incluso cuando la presentación aguda, la cistitis hemorrágica, se maneja con éxito (Ej: se detiene el sangrado), la disminución de la compliance vesical puede provocar efectos tardíos, incluidos síntomas urinarios y una función alterada del tracto inferior.53
La necesidad de derivación urinaria no es infrecuente en VP-RMS, ya sea por cistectomía inicial o tardía o por pérdida de la función de la vejiga relacionada con la radiación. Sin embargo, la buena calidad de vida puede ser restaurada a estos pacientes.54 Muchos autores abogan por el uso de una derivación urinaria incontinente inicial, a través de conductos ileales o colónicos, 55 mientras que otros han informado buenos resultados con la utilización inmediata de una derivacióncontinente ortotópica.37 Sin embargo, la derivación urinaria continente inicial conlleva un conjunto particular de desafíos: primero, cualquier complicación postoperatoria es probable que retrase la quimioterapia, y generalmente se acepta que la derivación continente tiene una tasa de complicaciones más alta que la incontinente.56 En segundo lugar, como se señaló anteriormente, la biopsia por congelación no es confiable para evaluar la idoneidad de los márgenes quirúrgicos.3740 Tercero, si la recurrencia local ocurre, una derivación urinaria continente hace que el tratamiento adecuado sea un gran desafío 57 En cuarto lugar, dado el rango de edad en el que se produce el VP-RMS, puede ser difícil evaluar si el niño y / o la familia son capaces de un mantenimiento adecuado a largo plazo de su desviación urinaria. Debido a estos desafíos específicos, muchos autores se expresan en contra de la reconstrucción inmediata, a menos que la terapia adicional sea altamente improbable y tanto el centro de tratamiento como el cirujano tengan amplia experiencia en reconstrucciones continentes.
Los pacientes con sustituciones intestinales (neovejiga, conducto o cistoplastia de aumento) tienen un mayor riesgo de anomalías metabólicas (hipocloremia, acidosis metabólica hipopotasémica con conductos ileales y de colon), cálculos y perforación. El uso del intestino en el tracto genitourinario también se asocia con un mayor riesgo de desarrollo de tumores malignos secundarios, con un riesgo que suele aumentar entre 5 y 10 años después de la cirugía.58 Los pacientes con desviaciones urinarias deben evaluarse al menos anualmente con imágenes y laboratorios del tracto superior (electrolitos, BUN y creatinina); Dado el curso temporal para el desarrollo de neoplasias malignas secundarias en esta población, la mayoría de los urólogos inician una cistoscopia anual y una citología urinaria de 5 a 7 años después de la derivación. Dado que el tratamiento para RMS se ha centrado cada vez más en la preservación de órganos, en la actualidad rara vez se emplean desviaciones urinarias.
Pronóstico
A diferencia de otros sitios GU, la localización en vejiga o próstata se considera un sitio desfavorable para RMS. Tiene un peor pronóstico que otros sitios y, el sistema de estadificación IRSG lo considera, en el mejor de los casos, como Estadío 2 antes del tratamiento. La sobrevida libre de eventos del VP-RMS en el ensayo IRS-IV fue del 77%. Los pacientes con enfermedad no metastásica tuvieron una sobrevida global del 82% a los 6 años. Sin embargo, hay grupos con pronósticos particularmente buenos después de un diagnóstico de VP-RMS. Se ha reportado que los niños con un tumor sarcoma botrioides endofítico tienen tasas de supervivencia a 10 años > 90%.40
Localización vulvar, vaginal, uterina
Epidemiología
Las neoplasias malignas ginecológicas son poco frecuentes en los niños y representan menos de 1 de cada 20 tumores infantiles. La mayoría de ellos son RMS vaginales.5960 Aunque el RMS en los sitios ginecológicos representa aproximadamente el 3,5% de todos los RMS pediátricos, 61 los estos tumores aún son extremadamente raros. Fernández reportó 18 pacientes en 39 años en una sola institución, 62 mientras que Kirsch encontró sólo 67 casos de RMS genitourinariopediátrico en niñas durante un periodo de 33 años.63 Si bien el RMS ginecológico puede ocurrir a cualquier edad, los tumores vaginales son más comunes en niñas pequeños y en edad preescolar (con un diagnóstico que se produce en una edad media de 3,7 años en un estudio).62 Existe una distribución bimodal de pacientes mujeres con RMS, con el primer pico entre 1 a 4 años de edad (en su mayoría tumores vaginales) y otro entre 15 a 19 años de edad (en su mayoría con localización primaria uterina / cervical).63 La mayoría de los pacientes (81%) son caucásicos, y una minoría es afroamericana (13%) o hispana (2%).61 En general, la vagina y el útero / cuello uterino son los sitios primarios más comunes, aunque las proporciones exactas varían según el estudio. Kirsch et al informaron que las proporciones eran relativamente iguales (42% en vagina en comparación con 39% en útero / cuello uterino), aunque una revisión de IRS I-IV encontró que la vagina era el sitio primario en el 54% de los pacientes, en comparación con 17% de los pacientes que lo presentaba en útero y 15% para el cuello uterino.6163 Dado que estos tumores son relativamente poco frecuentes, el error de muestreo podría explicar estas observaciones dispares.
Como en otros sitios, la histología del RMS ginecológico puede ser embrionario o alveolar. Los tumores uterinos y vaginales tienden a ser del Grupo III o IV en la presentación en la mayoría de los casos, dado el enfoque típico solo con una biopsia inicial, en comparación con los tumores vulvares, que generalmente son del Grupo I o II debido a la capacidad de ofrecer resección inicial. La gran mayoría (50-67%) de los tumores vaginales embrionarios son localizadosal momento del diagnóstico. En aquellos que son metastásicos, el pulmón es el sitio más común de enfermedad a distancia.64 Arndt informó un 45% de histología alveolar en tumores vulvares.61 Los tumores uterinos tendieron a ser grandes e invasivos (> 5 cm en la mayoría de los casos) y se asociaron con enfermedad nodal en 27%. En contraste, solo un paciente con RMS cervical invasivo fue identificado en la serie del IRS 61, y hasta la fecha, el RMS cervical nunca se ha asociado con enfermedad nodal.
Biología del tumor
El RMS es un tumor de células redondas pequeñas y azules con músculo esquelético diferenciado.65 Los tumores vaginales suelen tener dos subtipos: botrioide ("racimo de uvas") y embrionario. La aparición de botrioides surge secundariamente al crecimiento tumoral en el subepitelio de los órganos huecos, con la consiguiente distorsión de la capa submucosa de las células fusiformes.66 La variante alveolar del RMS es infrecuente en sitios vaginales / vulvares / uterinos, pero augura un peor pronóstico, excepto cuando se asocia con lesiones vulvares (que tienden a localizarse).67
Presentación clínica y estudios complementarios
El diagnóstico generalmente ocurre cuando los pacientes presentan síntomas, en lugar de ser hallazgos incidentales o detectados mediante screening. Los RMS vaginal y uterino se presentan con mayor frecuencia con flujo vaginal con sangre (67%) o una masa visible que se extiende desde la vagina (33%; Figura 3).596268 El diagnóstico diferencial de masas vulvares también incluye otras neoplasias malignas de la vulva (p. Ej., Tumor del seno endodérmico) o crecimientos vulvares benignos (p. Ej., Quistes del conducto de Gartner, canal de quistes de Nuck y hamartoma).69 En pacientes en los que la masa vulvar no se identifica fácilmente como neoplásica, debe considerarse un cariotipo, aunque, por supuesto, esto no debe retrasar el diagnóstico o el tratamiento en pacientes en los que se sospecha neoplasia. La imagen inicial puede consistir en un ultrasonido pélvico para delinear el tamaño y la extensión de la masa. Si se sospecha un proceso neoplásico, el estudio por método de corte del abdomen y la pelvis con una TC o RMN ayudará a determinar la extensión de la enfermedad local y ganglionar. Dado que los pulmones y la médula ósea son los sitios más comunes de enfermedad metastásica, el estudio metastásico inicial consiste en una tomografía computarizada del tórax, una biopsia de médula ósea y una gammagrafía ósea.
Tratamiento
Inicial - Diagnóstico
Históricamente (hasta 1972), el tratamiento consistió en exenteración pélvica, pero los protocolos IRS I / II (1972 y 1984, respectivamente) encontraron que la gran mayoría de los tumores eran exquisitamente sensibles a la quimioterapia.70 Siguiendo los protocolos del IRS I / II, sólo el 12% de los pacientes requirió exenteración pélvica, lo que significa que la preservación uterina podría lograrse en el 88% de los pacientes.71 En el IRS III / IV, solo el 22% de los pacientes se sometieron a histerectomía y el 82% estaban vivos 5 años después del diagnóstico.61 Al mismo tiempo, un protocolo de SIOP abierto entre 1984-1994 encontró una tasa de sobrevida superior al 90% con terapia multimodal y una tasa de preservación uterina del 88% entre los sobrevivientes (con dos pacientes fallecidos y tres pacientes que requirieron histerectomía).72 Sobre la base de estos hallazgos, las recomendaciones actuales son quimioterapia inicial seguida de escisión quirúrgica y, en algunos casos, radioterapia adyuvante.71 Por lo tanto, la exenteración pélvica está reservada para pacientes con tumores refractarios a la quimioterapia y la radiación.
Después de la quimioterapia, la exploración se realiza con escisión completa del tumor (cuando sea posible) y posiblemente otra biopsia para evaluar la viabilidad del tumor.61687374 El control local es primordial ya que la recurrencia local es responsable de la gran mayoría de las fallas de tratamiento. Un margen de 5 mm se considera suficiente para el control oncológico.74 Aunque se debe realizar una resección completa cuando sea posible (por ejemplo, los tumores vaginales pueden requerir histerectomía), los tumores que se extienden hacia la vejiga o el intestino deben resecarse por completo sólo cuando se puede preservar la función del órgano.74
Además de la evaluación por imágenes, los ganglios linfáticos también deben evaluarse por anatomía patológica. Las lesiones vulvares drenan a los ganglios linfáticos inguinales superficiales y profundos, mientras que los tumores vaginales y uterinos drenan a los ganglios linfáticos pélvicos. Las complicaciones quirúrgicas se analizan con más detalle a continuación, e incluyen la fístula rectovaginal o vesicovaginal y la incontinencia urinaria, por lo general en pacientes con resecciones extensas, incluida la exenteración pélvica.62
Re-resección pretratamiento
La re-resección previa al tratamiento se emplea típicamente en pacientes en los que se resecó una masa "completamente" (es decir, no se realizó biopsia) antes del diagnóstico de RMS. La evaluación histológica de RMS en estos casos a menudo revela márgenes positivos y resección incompleta de la masa. Antes de administrar la terapia adyuvante, es apropiada la re-resección de la masa con la eliminación de cualquier tejido residual sospechoso con un amplio margen local. El muestreo de ganglios linfáticos, si no se realiza en el momento de la cirugía inicial, también debe realizarse en este momento. Al igual que con la cirugía después de la quimioterapia y la radioterapia, debe evitarse la escisión de órganos, como la vejiga, que puede tener consecuencias funcionales significativas para el paciente.74
Operación de Second Look (SLO)
Las operaciones de Second Look se realizan periódicamente para evaluar la carga tumoral y la respuesta clínica después de la radioterapia y la quimioterapia. La extirpación del tumor se puede realizar si da lugar a una disminución de la terapia adicional (por ejemplo, radiación). Debido a que el RMS vaginal y vulvar suele ser altamente sensible a la quimioterapia, los SLO rara vez se realizan para los tumores en estos sitios.74
Tratamiento adyuvante - Quimioterapia / Radioterapia
El régimen estándar de quimioterapia para el RMS es VAC. Los pacientes que no recibieron Adriamcina tuvieron una tasa de recurrencia del 15%, en comparación con la curación completa en los pacientes que si la recibieron. La ifosfamida se sustituye ocasionalmente por ciclofosfamida. La radiación se utiliza para pacientes en los que hay tumor residual después de la quimioterapia. Históricamente, la dosis ha sido 5040cGy para pacientes con enfermedad grave y 4140cGy para pacientes con enfermedad microscópica. Un estudio reciente del COG (D9602) se propuso determinar si se podrían lograr resultados de sobrevida comparables a los observados en el IRS III / IV con dosis de radioterapia de haz externo más bajas (3600 cGy en pacientes con márgenes microscópicos positivos, 4140 cGy en pacientes con enfermedad nodal y 5040 cG en pacientes con enfermedad residual gruesa).74 Las niñas con tumores vaginales representaron la mayoría de los fracasos del tratamiento, con control local logrado en solo la mitad de este subgrupo, aunque muchas no recibieron agentes alquilantes ni radioterapia, lo que hace que la razón del fracaso sea difícil de determinar con certeza. Los protocolos actuales del COG exigen la radioterapia para los tumores vaginales de los grupos II y III, con una intensidad de radioterapia que varía de 3600-5040 cGy según la extensión del tumor residual. En estos pacientes, la tasa de fracaso local aumenta sin radiación.75
El fracaso de la radiación parece ser más pronunciado en niños menores de 3 años, posiblemente debido a una dosis inadecuada relacionada con el mayor temor a las complicaciones tardías relacionadas con la radiación, así como a una menor comprensión y cooperación con los objetivos de la terapia.7677 No obstante, el fallo de la radiación parece aumentar inequívocamente el riesgo de recurrencia (70%) y muerte. Aunque no se usa de manera rutinaria en los Estados Unidos, la braquiterapia para el RMS pediátrico vaginal y vulvar se ha empleado en Europa.7879 Magne et al. informaron una supervivencia alta (91%) a cinco años en 32 pacientes que recibieron braquiterapia por enfermedad residual, y sólo dos experimentaron efectos secundarios agudos inducidos por radiación. Sin embargo, la tasa de estenosis vaginal en este grupo fue del 20% en 8 años.
Pronóstico y seguimiento
Los factores pronósticos que acompañan a una mejor tasa de sobrevida incluyen la edad joven (<10 años), la enfermedad confinada localmente y el tratamiento quimioterapéutico con VAC y (cuando sea apropiado) la radioterapia.61 La vulva y la vagina se consideran sitios favorables. Los tumores mayores de 5 cm, o aquellos asociados con invasión local o ganglios linfáticos positivos, se asociaron con resultados más pobres. Los niños mayores de un año tuvieron un mejor pronóstico que los bebés, probablemente secundario a la capacidad de tolerar las complicaciones relacionadas con la terapia.380 Casi todas las muertes ocurrieron en los primeros 5 años después del diagnóstico, pero se ha informado posibilidad de recaída tardía.81
Efectos tardíos
Como el tratamiento de RMS se centra en la preservación de órganos, los efectos tardíos se centran cada vez más en las secuelas de la quimioterapia y la radioterapia en lugar de la cirugía. Spunt et al observaron múltiples quejas en pacientes tratados por RMS, incluidos problemas reproductivos (dispareunia, estenosis vaginal, fístula vaginal, efectos endócrinos, ciclos menstruales anormales, menopausia prematura), trastornos genitourinarios (función vesical anormal, obstrucción ureteral, pielonefritis, obstrucción uretral) y alteración de la función gastrointestinal (estenosis, fístula, perforación, colecistitis, gastritis, incontinencia fecal).82 Aunque esta lista es extensa, los autores advirtieron que el desarrollo tardío de muchas complicaciones (particularmente en pacientes que reciben radiación) puede significar que se subestima la verdadera prevalencia de los efectos tardíos. Además, en la actualidad no existen métodos estandarizados para evaluar la integridad y la función del piso pélvico en los sobrevivientes de RMS, por lo que las quejas sutiles de vejiga e intestino pueden no ser reportadas.
Si bien la función gonadal endócrina es la más comúnmente afectada (p. Ej., Insuficiencia ovárica prematura, especialmente con radiación pélvica), las complicaciones más comunes que requieren intervención quirúrgica fueron las afecciones ginecológicas, como estenosis y fístulas. Las fístulas se asociaron universalmente con la radioterapia, con una proporción igual de espontánea y postquirúrgica. La estenosis vaginal generalmente se puede tratar con dilatación o una reparación quirúrgica más formal; los riesgos de este último incluyen la perforación intestinal y la estenosis recurrente, por lo que la reparación se realiza normalmente solo cuando el manejo conservador ha fallado o tiene una probabilidad extremadamente baja de éxito inicial.
Otros sistemas en los que se han observado efectos tardíos incluyen el sistema musculoesquelético (escoliosis y fibrosis tisular), problemas de salud mental (depresión, ansiedad, insomnio, problemas académicos, abuso de sustancias) y cardiovasculares (derrame cerebral, hipertensión, cardiomiopatía asociada a doxorubicina). Las neoplasias secundarias tienen una incidencia acumulada del 13,5% a los 20 años. Spunt et al. informaron sobre tres pacientes, todos los cuales desarrollaron tumores malignos secundarios (osteosarcoma, carcinoma de células escamosas in situ del cuello uterino y carcinoma de colon) dentro del campo de radiación de RMS. En esta serie, la proporción de pacientes que desarrollaron neoplasias secundarias fue mayor que la de otros grupos de sobrevivientes de cáncer, aunque no estaba claro si este hallazgo era falso dado el pequeño número de pacientes afectados, o representativo de un riesgo verdaderamente mayor dada la recepción casi universal de radioterapia en esa cohorte. La probabilidad de desarrollar un efecto tardío no se correlacionó con la edad en el momento del diagnóstico de RMS. Los autores encontraron que los refinamientos en la terapia para RMS resultaron en más efectos tardíos para los pacientes tratados después de 1984 que en los anteriores, pero esto se debió solo a un aumento en el número de toxicidad de grado inferior (1 y 2).
La radioterapia es quizás el factor de riesgo más importante para el desarrollo de complicaciones tardías. En el estudio mencionado anteriormente de 26 pacientes por Spunt et al, sólo dos no tuvieron efectos tardíos, ambos de los cuales no habían recibido radiación. Los pacientes afectados tuvieron un promedio de 4.5 efectos tardíos, con un máximo de 14 por paciente; los pacientes con radiación tuvieron una mediana de 9.5 efectos tardíos cada uno, en comparación con un efecto tardío en pacientes que no habían recibido radiación. Los efectos tardíos también fueron más graves en los pacientes que se habían sometido a radioterapia, con efectos más graves (grado 3 a 4) informados en este grupo. Más de la mitad (54%) se sometieron a cirugía para controlar las complicaciones, siendo la dilatación vaginal la más común, seguida de procedimientos para corregir las estenosis gastrointestinales y las fístulas vaginales. Los autores concluyeron que la radiación confirió un mayor riesgo de complicaciones tardías relacionadas con el tratamiento en niños que en adultos. Dada la estrecha relación anatómica en la pelvis entre las estructuras ginecológicas y la vejiga, muchos de los efectos tardíos de la misma vejiga se pueden ver como en el VP-RMS que se revisaron anteriormente.
Infertilidad
Aunque se han informado problemas de fertilidad después del tratamiento para muchos cánceres infantiles, la literatura específica sobre problemas de fertilidad después del tratamiento para RMS vaginal / vulvar / uterino sigue siendo escasa. La fertilidad puede ser desafiada debido a la exposición a la quimioterapia, la radioterapia y la intervención quirúrgica. La insuficiencia ovárica prematura, como ya se mencionó, es un hallazgo común después de la radiación pélvica. En pacientes que se han sometido a una histerectomía, se pierde la capacidad de llevar a un niño. Incluso en aquellos en los que se conserva todo o parte del útero, los cambios posquirúrgicos o posradiación pueden promover la incompetencia cervical o el parto prematuro. Sin embargo, se han reportado nacimientos vivos después del tratamiento para RMS pélvico.83 Las tasas de embarazo después del tratamiento para el sarcoma son moderadas (47%) en al menos otro estudio 84, lo que sugiere que la radiación pélvica y las secuelas quirúrgicas son predictores más fuertes de infertilidad que la quimioterapia en las niñas.
En los hombres que han recibido quimioterapia, se han informado problemas de infertilidad anteriormente. Un estudio describió una tasa de paternidad del 24%, con la mayoría de los otros pacientes con oligo o azoospermia.84 Los pacientes con paternidad exitosa no tuvieron exposición a quimioterapia alquilante o radiación pélvica. Otro estudio encontró análisis de semen universalmente anormales en hombres expuestos a ciclofosfamida antes de la pubertad.85 El grado de oligo o azoospermia no fue dependiente de la dosis en este estudio. Si bien la mayoría de los pacientes no tenían anomalías del eje hipotálamo-hipófisis-gonadal asociadas con los análisis anormales del semen, el 40% de los niños tenía niveles elevados de hormona luteinizante a pesar de la testosterona normal, lo que sugiere una función anormal de las células de Leydig. Es posible que la ifosfamida cause menos problemas de fertilidad que la ciclofosfamida.8687
Localizaciones primarias infrecuentes
Pélvica
El RMS que no surge de uno de los órganos genitourinarios es infrecuente. En la presentación, estos tumores a menudo son grandes y extensos, y encierran estructuras pélvicas, como nervios y vasos sanguíneos. Al igual que con el RMS que surge claramente de un órgano, el tratamiento apunta a optimizar la posibilidad de una escisión completa y minimizar el daño a otros órganos y estructuras pélvicas.88
Renal
El RMN renal es raro y en un período de 33 años, el grupo IRS multicéntrico incluyó sólo 10 sarcomas renales y, específicamente, sólo seis casos de RMS primaria renal.89 Por lo tanto, un diagnóstico rápido y preciso requiere un alto índice de sospecha y puede ser un desafío. Tanto el tumor de Wilms como el carcinoma de células renales deben excluirse del diagnóstico diferencial. La miogenina y MyoD1 son marcadores de músculo esquelético que se consideran tanto sensibles como específicos para el diagnóstico de RMS, ya que ambos se expresan temprano en la diferenciación del músculo esquelético.90 Si bien puede verse la positividad de WT1, típicamente no se ve un patrón de tinción nuclear en RMS como en Tumor de Wilms.91 La desmina es también un marcador para RMS.92
Una revisión retrospectiva del RMS primario renal del IRSG demostró que los tumores eran generalmente grandes (el diámetro medio oscilaba entre 8 y 15 cm), aunque la mayoría no eran invasivos. El RMS renal tuvo una alta incidencia de anaplasia (67%), mucho más alta que la reportada en otros tumores renales infantiles o en el RMS en general. Uno de los seis pacientes tenía enfermedad nodal y dos pacientes presentaron metástasis. En esta serie, todos los tumores fueron del subtipo embrionario.89
Al igual que con el RMS en otras ubicaciones primarias, la extensión de la escisión del RMS renal parece correlacionarse con la supervivencia. El COG recomienda la resección primaria completa del tumor cuando sea posible, incluida la disección de ganglios linfáticos regionales para la estadificación de la enfermedad. El RMS renal se considera un sitio desfavorable. En la serie IRSG de 6 pacientes, el tratamiento inicial consistió en una nefrectomía radical seguida de quimioterapia con 3 fármacos (VAC) en la mayoría de los casos, con radiación reservada para pacientes con enfermedad residual. Tres de seis pacientes presentaron enfermedad recurrente en el cerebro o en el hueso y murieron de la enfermedad. Los otros tres estaban vivos y sin enfermedad evaluable más de una década después de completar la terapia.89 En la actualidad, se recomienda una biopsia seguida de quimioterapia por adelantado para facilitar la extracción del tumor y preservar al máximo los órganos afectados y adyacentes en el momento de la cirugía. Después de la quimioterapia, la radioterapia puede emplearse en pacientes con tumor residual (márgenes positivos o enfermedad ganglionar) si los beneficios superan los riesgos, incluso en presencia de una anatomía abdominal anormal.93 Sin embargo, en la serie IRSG, incluso con radioterapia adyuvante, a los pacientes con enfermedad ganglionar o metástasis les fue mal.
Retroperitoneal
El RMS retroperitoneal como sitio primario es raro. En conjunto, el RMS pélvico retroperitoneal y no genitourinario representa solo el 10% de todo los RMS94 y en una serie institucional, sólo se registraron siete RMS retroperitonealesprimarios en un período de 30 años.95 Los tumores suelen identificarse después de que el paciente presenta síntomas (a menudo inespecíficos, como dolor o molestias abdominales) y, por lo tanto, muchos tumores son bastante grandes (la mayoría tienen más de 5 cm de diámetro en el momento del diagnóstico). Los RMS retroperitoneales primarios también tienden a ser más extensos a nivel local. Pham et al. informaron que sólo alrededor de un tercio de los pacientes tenían una enfermedad completamente localizada: el 62% tenía tumores que afectaban a los órganos cercanosy, aunque ninguno tenía metástasis a distancia en el momento del diagnóstico, se encontró que el 14% tenía enfermedad metastásica durante el tratamiento.95 Blakely et al. observó que el 94% de los tumores eran del grupo clínico III o IVy, en los niños en los que se registró el grado de invasión, el 96% tenía tumores invasivos.94
La mayoría de los tumores se encuentran en el nivel toracolumbar, con una minoría significativa en el nivel lumbosacro.95 Los varones y los niños menores de 5 años fueron los grupos más afectados. En estos grupos, predominó la histología embrionaria, mientras que la histología alveolar fue más común en niños mayores (por ejemplo, adolescentes). Los factores predictivos para mejoría libre de eventos y la sobrevida general fueron la edad más joven en el momento del diagnóstico, el sexo femenino y la histología embrionaria. Los pacientes con histología embrionaria fueron más propensos a sufrir una reducción tumoral en comparación con los niños con histología alveolar (que generalmente se sometieron a biopsia sola). La resección quirúrgica, incluso cuando el tumor no fue resecado por completo, se correlacionó con una mejor sobrevida. Mientras que los pacientes con RMS tuvieron una sobrevida sustancialmente más corta que los pacientes con otros tipos de sarcoma, la sobrevida global a cinco años fue similar en pacientes con y sin RMS.
Tabla 1A. Estadificación TNM
| Tumor | | |
|---|---|---|
| T1 | Confinado al sitio anatómico de origen | |
| T1a | ≤ 5 cm de diámetro en tamaño | |
| T1b | > 5 cm de diámetro en tamaño | |
| T2 | Extensión y / o fijación al tejido circundante | |
| T2a | ≤ 5 cm de diámetro en tamaño | |
| T2b | > 5 cm de diámetro en tamaño | |
| Nodos | | |
| N0 | Ganglios regionales sin afectación clínica | |
| N1 | Ganglios regionales CON afectación clínica | |
| Nx | Se desconoce el estado clínico de los nodos regionales (especialmente los sitios que impiden la evaluación) | |
| Metástasis | | |
| M0 | No hay metástasis a distancia | |
| M1 | Metástasis presentes | |
Tabla 1B. Estadificación IRSG
| Estadío | Sitio Anatómico | T | N | M | |
|---|---|---|---|---|---|
| 1 | Vaginal / uterino / cervical y paratesticular | cualquiera | cualquiera | M0 | |
| 2 | Vejiga / próstata | T1a / T2a (≤5cm) | N0 o Nx | M0 | |
| 3 | Vejiga / próstata | T1a / T2a (≤5cm) o T1b / T2b (> 5cm) | N1 cualquiera | M0 | |
| 4 | cualquiera | cualquiera | cualquiera | M1 | |
Tabla 2. Agrupación clínica posquirúrgica
| Grupo | | |
|---|---|---|
| I | Enfermedad localizada, completamente resecada (márgenes microscópicamente negativos) y nodos regionales no involucrados La biopsia o el muestreo de ganglios linfáticos es altamente recomendable | |
| I a | Confinado al órgano de origen. | |
| Ib | Compromiso de estructuras contiguas: infiltración fuera del músculo u órgano de origen, a través de los planos fasciales | |
| II | Resección total macroscópica con evidencia de diseminación regional. | |
| IIa | Tumor con resección macroscópica pero con enfermedad residual microscópica, sin afectación ganglionar regional | |
| IIb | Enfermedad regional con ganglios afectados, pero resecada completamente sin residuo microscópicos | |
| IIc | Enfermedad regional con ganglios afectados, resecada en gran medida, pero con evidencia de afectación microscópica residual y / o histológica del ganglio regional más distal en la disección | |
| III | Resección incompleta con enfermedad residual macroscópica | |
| IIIa | Después de la biopsia solamente | |
| IIIb | Después de la resección de la lesión primaria (> 50%) | |
| IV | Enfermedad metastásica distante presente al inicio | |
Table 3A. Clasificación de riesgo
| Riesgo | Histología | Grupo postoperatorio | Etapa IRSG Pre-Op | Años |
|---|---|---|---|---|
| Bajo | Embrionario | I, II, III | 1 | Todos |
| Embrionario | Yo, yo | 2, 3 | Todos | |
| Intermedio | ||||
| Embrionario | III | 2, 3 | Todos | |
| Alveolar | I, II, III | 1, 2, 3 | Todos | |
| Alto | Embrionario | IV | 4 | Todos |
| Alveolar | IV | 4 | Todos |
Table 3B. Clasificación de riesgo Clasificación de riesgo específica según el sitio de localización GU
| Sitio primario | Etapa Pre-Op | Enfoque quirúrgico inicial | Grupo postoperatorio | Histología | Años | Riesgo |
|---|---|---|---|---|---|---|
| Paratesticular | 1 | Orquiectomia | I, II, III | Embrionario | Todos | Bajo |
| 1 | Orquiectomia | I, II, III | Alveolar | Todos | Intermedio | |
| 4 | Orquiectomia | IV | Embrionario | Todos | Alto | |
| 4 | Orquiectomia | IV | Alveolar | Todos | Alto | |
| Vaginal / uterino / cervical | 1 | Resección | I, II, III | Embrionario | Todos | Bajo |
| 1 | Biopsia | III | Embrionario | Todos | Bajo | |
| 1 | Resección | I, II, III | Alveolar | Todos | Intermedio | |
| 1 | Biopsia | III | Alveolar | Todos | Intermedio | |
| 4 | Resección o biopsia | IV | Embrionario | Todos | Alto | |
| 4 | Resección o biopsia | IV | Alveolar | Todos | Alto | |
| Vejiga / próstata | 2 o 3 | Resección | Yo o II | Embrionario | Todos | Bajo |
| 2 o 3 | Resección | III | Embrionario | Todos | Intermedio | |
| 2 o 3 | Biopsia | III | Embrionario | Todos | Intermedio | |
| 2 o 3 | Resección | I, II, III | Alveolar | Todos | Intermedio | |
| 2 o 3 | Biopsia | III | Alveolar | Todos | Intermedio | |
| 4 | Resección o biopsia | IV | Embrionario | Todos | Alto | |
| 4 | Resección o biopsia | IV | Alveolar | Todos | Alto |
Clave de abreviaturas:
Rabdomiosarcoma (RMS) Embrionario Rhadomyosarcoma (E-RMS) Botrioide Rabdomiosarcoma (B-RMS) Alveolar Rabdomiosarcoma (A-RMS) Vejiga y / o de la Próstata (V / P) Genitourinario (GU) Inmunohistoquímica (IHC) Pérdida de heterocigosidad (LOH) Intergroup Rhabdomyosarcoma Study Group (IRSG) Children's Oncology Group (COG) Disección retroperitoneal de ganglios linfáticos (RPLND) Paratesticular (PT) Vincristine, Actinomycin-D, and Cyclophosphamide (VAC) Tumor-Nodulos-Metastasis (TNM) Nodos linfáticos (LN) ) Supervivencia global (OS) Supervivencia libre de eventos (EFS)
Referencias
-
Dasgupta, R., Rodeberg, D. A.: Update on rhabdomyosarcoma. Semin Pediatr Surg, 21: 68, 2012 ↩ ↩2 ↩3
-
Crist, W., Gehan, E. A., Ragab, A. H. et al.: The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol, 13: 610, 1995 ↩
-
Meza, J. L., Anderson, J., Pappo, A. S. et al.: Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children's Oncology Group. J Clin Oncol, 24: 3844, 2006 ↩ ↩2 ↩3
-
Pappo, A. S., Shapiro, D. N.: Rhabdomyosarcoma: biology and therapy. Cancer Treat Res, 92: 309, 1997 ↩
-
Perez, E. A., Kassira, N., Cheung, M. C. et al.: Rhabdomyosarcoma in children: a SEER population based study. J Surg Res, 170: e243, 2011 ↩
-
Ferrer, F. A., Isakoff, M., Koyle, M. A.: Bladder/prostate rhabdomyosarcoma: past, present and future. J Urol, 176: 1283, 2006 ↩
-
Joshi, D., Anderson, J. R., Paidas, C. et al.: Age is an independent prognostic factor in rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. Pediatr Blood Cancer, 42: 64, 2004 ↩
-
Malempati, S., Hawkins, D. S.: Rhabdomyosarcoma: review of the Children's Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer, 59: 5, 2012 ↩
-
Sung, L., Anderson, J. R., Arndt, C. et al.: Neurofibromatosis in children with Rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma study IV. J Pediatr, 144: 666, 2004 ↩
-
Malkin, D., Li, F. P., Strong, L. C. et al.: Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science, 250: 1233, 1990 ↩
-
Ruymann, F. B., Maddux, H. R., Ragab, A. et al.: Congenital anomalies associated with rhabdomyosarcoma: an autopsy study of 115 cases. A report from the Intergroup Rhabdomyosarcoma Study Committee (representing the Children's Cancer Study Group, the Pediatric Oncology Group, the United Kingdom Children's Cancer Study Group, and the Pediatric Intergroup Statistical Center). Med Pediatr Oncol, 16: 33, 1988 ↩
-
Asmar, L., Gehan, E. A., Newton, W. A. et al.: Agreement among and within groups of pathologists in the classification of rhabdomyosarcoma and related childhood sarcomas. Report of an international study of four pathology classifications. Cancer, 74: 2579, 1994 ↩
-
Dias, P., Chen, B., Dilday, B. et al.: Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol, 156: 399, 2000 ↩
-
Heerema-McKenney, A., Wijnaendts, L. C., Pulliam, J. F. et al.: Diffuse myogenin expression by immunohistochemistry is an independent marker of poor survival in pediatric rhabdomyosarcoma: a tissue microarray study of 71 primary tumors including correlation with molecular phenotype. Am J Surg Pathol, 32: 1513, 2008 ↩
-
Gordon, T., McManus, A., Anderson, J. et al.: Cytogenetic abnormalities in 42 rhabdomyosarcoma: a United Kingdom Cancer Cytogenetics Group Study. Med Pediatr Oncol, 36: 259, 2001 ↩
-
Anderson, J., Gordon, T., McManus, A. et al.: Detection of the PAX3-FKHR fusion gene in paediatric rhabdomyosarcoma: a reproducible predictor of outcome? Br J Cancer, 85: 831, 2001 ↩ ↩2
-
Sorensen, P. H., Lynch, J. C., Qualman, S. J. et al.: PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children's oncology group. J Clin Oncol, 20: 2672, 2002 ↩
-
Missiaglia, E., Williamson, D., Chisholm, J. et al.: PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol, 30: 1670, 2012 ↩
-
Davicioni, E., Anderson, M. J., Finckenstein, F. G. et al.: Molecular classification of rhabdomyosarcoma–genotypic and phenotypic determinants of diagnosis: a report from the Children's Oncology Group. Am J Pathol, 174: 550, 2009 ↩ ↩2
-
El-Badry, O. M., Minniti, C., Kohn, E. C. et al.: Insulin-like growth factor II acts as an autocrine growth and motility factor in human rhabdomyosarcoma tumors. Cell Growth Differ, 1: 325, 1990 ↩
-
Williamson, D., Lu, Y. J., Gordon, T. et al.: Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J Clin Oncol, 23: 880, 2005 ↩
-
Lawrence, W., Jr., Anderson, J. R., Gehan, E. A. et al.: Pretreatment TNM staging of childhood rhabdomyosarcoma: a report of the Intergroup Rhabdomyosarcoma Study Group. Children's Cancer Study Group. Pediatric Oncology Group. Cancer, 80: 1165, 1997 ↩
-
Stewart, R. J., Martelli, H., Oberlin, O. et al.: Treatment of children with nonmetastatic paratesticular rhabdomyosarcoma: results of the Malignant Mesenchymal Tumors studies (MMT 84 and MMT 89) of the International Society of Pediatric Oncology. J Clin Oncol, 21: 793, 2003 ↩ ↩2
-
Wiener, E. S., Anderson, J. R., Ojimba, J. I. et al.: Controversies in the management of paratesticular rhabdomyosarcoma: is staging retroperitoneal lymph node dissection necessary for adolescents with resected paratesticular rhabdomyosarcoma? Semin Pediatr Surg, 10: 146, 2001 ↩ ↩2
-
Wiener, E. S., Lawrence, W., Hays, D. et al.: Retroperitoneal node biopsy in paratesticular rhabdomyosarcoma. J Pediatr Surg, 29: 171, 1994 ↩
-
Ferrari, A., Bisogno, G., Casanova, M. et al.: Paratesticular rhabdomyosarcoma: report from the Italian and German Cooperative Group. J Clin Oncol, 20: 449, 2002 ↩ ↩2 ↩3 ↩4
-
Ferrari, A., Bisogno, G., Casanova, M. et al.: Is alveolar histotype a prognostic factor in paratesticular rhabdomyosarcoma? The experience of Italian and German Soft Tissue Sarcoma Cooperative Group. Pediatr Blood Cancer, 42: 134, 2004 ↩ ↩2 ↩3
-
Federico, S. M., Spunt, S. L., Krasin, M. J. et al.: Comparison of PET-CT and conventional imaging in staging pediatric rhabdomyosarcoma. Pediatr Blood Cancer, 60: 1128, 2013 ↩
-
Burnette, J. O., Klaassen, Z., Hatley, R. M. et al.: Staging Paratesticular Rhabdomyosarcoma in the "as Low as Reasonably Achievable" Age: The Case for PET-CT. Urology, 2013 ↩
-
Crist, W. M., Anderson, J. R., Meza, J. L. et al.: Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol, 19: 3091, 2001 ↩ ↩2
-
Tomaszewski, J. J., Sweeney, D. D., Kavoussi, L. R. et al.: Laparoscopic retroperitoneal lymph node dissection for high-risk pediatric patients with paratesticular rhabdomyosarcoma. J Endourol, 24: 31, 2010 ↩
-
Cost, N. G., Dajusta, D. G., Granberg, C. F. et al.: Robot-Assisted Laparoscopic Retroperitoneal Lymph Node Dissection in an Adolescent Population. J Endourol, 2012 ↩
-
Meir, D. B., Inoue, M., Gur, U. et al.: Urinary diversion in children with pelvic tumors. J Pediatr Surg, 39: 1787, 2004 ↩ ↩2
-
Hays, D. M., Raney, R. B., Jr., Lawrence, W., Jr. et al.: Bladder and prostatic tumors in the intergroup Rhabdomyosarcoma study (IRS-I): results of therapy. Cancer, 50: 1472, 1982 ↩
-
Lerner, S. P., Hayani, A., O'Hollaren, P. et al.: The role of surgery in the management of pediatric pelvic rhabdomyosarcoma. J Urol, 154: 540, 1995 ↩
-
Arndt, C., Rodeberg, D., Breitfeld, P. P. et al.: Does bladder preservation (as a surgical principle) lead to retaining bladder function in bladder/prostate rhabdomyosarcoma? Results from intergroup rhabdomyosarcoma study iv. J Urol, 171: 2396, 2004 ↩ ↩2 ↩3 ↩4 ↩5 ↩6
-
Merguerian, P. A., Agarwal, S., Greenberg, M. et al.: Outcome analysis of rhabdomyosarcoma of the lower urinary tract. J Urol, 160: 1191, 1998 ↩ ↩2 ↩3
-
Chui, C. H., Spunt, S. L., Liu, T. et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg, 37: 1424, 2002 ↩
-
Heyn, R., Newton, W. A., Raney, R. B. et al.: Preservation of the bladder in patients with rhabdomyosarcoma. J Clin Oncol, 15: 69, 1997 ↩
-
Leuschner, I., Harms, D., Mattke, A. et al.: Rhabdomyosarcoma of the urinary bladder and vagina: a clinicopathologic study with emphasis on recurrent disease: a report from the Kiel Pediatric Tumor Registry and the German CWS Study. Am J Surg Pathol, 25: 856, 2001 ↩ ↩2 ↩3
-
Stevens, M. C., Rey, A., Bouvet, N. et al.: Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology–SIOP Malignant Mesenchymal Tumor 89. J Clin Oncol, 23: 2618, 2005 ↩ ↩2
-
Yeung, C. K., Ward, H. C., Ransley, P. G. et al.: Bladder and kidney function after cure of pelvic rhabdomyosarcoma in childhood. Br J Cancer, 70: 1000, 1994 ↩ ↩2
-
Ozkan, K. U., Bauer, S. B., Khoshbin, S. et al.: Neurogenic bladder dysfunction after sacrococcygeal teratoma resection. J Urol, 175: 292, 2006 ↩
-
Mosiello, G., Gatti, C., De Gennaro, M. et al.: Neurovesical dysfunction in children after treating pelvic neoplasms. BJU Int, 92: 289, 2003 ↩
-
Cruccetti, A., Kiely, E. M., Spitz, L. et al.: Pelvic neuroblastoma: low mortality and high morbidity. J Pediatr Surg, 35: 724, 2000 ↩
-
Ritchey, M., Ferrer, F., Shearer, P. et al.: Late effects on the urinary bladder in patients treated for cancer in childhood: a report from the Children's Oncology Group. Pediatr Blood Cancer, 52: 439, 2009 ↩
-
Heyn, R., Raney, R. B., Jr., Hays, D. M. et al.: Late effects of therapy in patients with paratesticular rhabdomyosarcoma. Intergroup Rhabdomyosarcoma Study Committee. J Clin Oncol, 10: 614, 1992 ↩
-
Hale, G. A., Marina, N. M., Jones-Wallace, D. et al.: Late effects of treatment for germ cell tumors during childhood and adolescence. J Pediatr Hematol Oncol, 21: 115, 1999 ↩
-
Dorr, W., Beck-Bornholdt, H. P.: Radiation-induced impairment of urinary bladder function in mice: fine structure of the acute response and consequences on late effects. Radiat Res, 151: 461, 1999 ↩
-
Tefft, M., Lattin, P. B., Jereb, B. et al.: Acute and late effects on normal tissues following combined chemo- and radiotherapy for childhood rhabdomyosarcoma and Ewing's sarcoma. Cancer, 37: 1201, 1976 ↩
-
Mangar, S. A., Foo, K., Norman, A. et al.: Evaluating the effect of reducing the high-dose volume on the toxicity of radiotherapy in the treatment of bladder cancer. Clin Oncol (R Coll Radiol), 18: 466, 2006 ↩ ↩2
-
Hays, D. M., Raney, R. B., Wharam, M. D. et al.: Children with vesical rhabdomyosarcoma (RMS) treated by partial cystectomy with neoadjuvant or adjuvant chemotherapy, with or without radiotherapy. A report from the Intergroup Rhabdomyosarcoma Study (IRS) Committee. J Pediatr Hematol Oncol, 17: 46, 1995 ↩
-
Jerkins, G. R., Noe, H. N., Hill, D.: Treatment of complications of cyclophosphamide cystitis. J Urol, 139: 923, 1988 ↩
-
Filipas, D., Fisch, M., Stein, R. et al.: Rhabdomyosarcoma of the bladder, prostate or vagina: the role of surgery. BJU Int, 93: 125, 2004 ↩
-
Duel, B. P., Hendren, W. H., Bauer, S. B. et al.: Reconstructive options in genitourinary rhabdomyosarcoma. J Urol, 156: 1798, 1996 ↩
-
van Hemelrijck, M., Thorstenson, A., Smith, P. et al.: Risk of in-hospital complications after radical cystectomy for urinary bladder carcinoma: population-based follow-up study of 7608 patients. BJU Int, 112: 1113, 2013 ↩
-
Castagnetti, M., Angelini, L., Alaggio, R. et al.: Oncological outcome and urinary function after radical cystectomy for rhabdomyosarcoma in children: role of the orthotopic ileal neo-bladder based on a 15-year experience at a single centre. J Urol, In press , 2013 ↩
-
Austen, M., Kalble, T.: Secondary malignancies in different forms of urinary diversion using isolated gut. J Urol, 172: 831, 2004 ↩
-
Hassan, E., Creatsas, G., Michalas, S.: Genital tumors during childhood and adolescence. A clinical and pathological study of 71 cases. Clin Exp Obstet Gynecol, 26: 20, 1999 ↩ ↩2
-
Hellman, K., Silfversward, C., Nilsson, B. et al.: Primary carcinoma of the vagina: factors influencing the age at diagnosis. The Radiumhemmet series 1956-96. Int J Gynecol Cancer, 14: 491, 2004 ↩
-
Arndt, C. A., Donaldson, S. S., Anderson, J. R. et al.: What constitutes optimal therapy for patients with rhabdomyosarcoma of the female genital tract? Cancer, 91: 2454, 2001 ↩ ↩2 ↩3 ↩4 ↩5 ↩6 ↩7 ↩8
-
Fernandez-Pineda, I., Spunt, S. L., Parida, L. et al.: Vaginal tumors in childhood: the experience of St. Jude Children's Research Hospital. J Pediatr Surg, 46: 2071, 2011 ↩ ↩2 ↩3 ↩4
-
Kirsch, C. H., Goodman, M., Esiashvili, N.: Outcome of Female Pediatric Patients Diagnosed With Genital Tract Rhabdomyosarcoma Based on Analysis of Cases Registered in SEER Database Between 1973 and 2006. Am J Clin Oncol, 37: 47, 2014 ↩ ↩2 ↩3
-
Copeland, L. J., Sneige, N., Stringer, C. A. et al.: Alveolar rhabdomyosarcoma of the female genitalia. Cancer, 56: 849, 1985 ↩
-
Qualman, S. J., Coffin, C. M., Newton, W. A. et al.: Intergroup Rhabdomyosarcoma Study: update for pathologists. Pediatr Dev Pathol, 1: 550, 1998 ↩
-
Newton, W. A., Jr., Gehan, E. A., Webber, B. L. et al.: Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification–an Intergroup Rhabdomyosarcoma Study. Cancer, 76: 1073, 1995 ↩
-
Scarpato, K. R., Ferrer, F. A.: Genitourinary rhabdomyosarcoma in children. In: AUA Update Series, vol. 32 (9), 2013 ↩
-
Grimsby, G. M., Ritchey, M. L.: Pediatric urologic oncology. Pediatr Clin North Am, 59: 947, 2012 ↩ ↩2
-
Youngstrom, E. A., Bartkowski, D. P.: Vulvar embryonal rhabdomyosarcoma: a case report. J Pediatr Urol, 9: e144, 2013 ↩
-
Hays, D. M., Shimada, H., Raney, R. B., Jr. et al.: Sarcomas of the vagina and uterus: the Intergroup Rhabdomyosarcoma Study. J Pediatr Surg, 20: 718, 1985 ↩
-
Andrassy, R. J., Hays, D. M., Raney, R. B. et al.: Conservative surgical management of vaginal and vulvar pediatric rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma Study III. J Pediatr Surg, 30: 1034, 1995 ↩ ↩2
-
Martelli, H., Oberlin, O., Rey, A. et al.: Conservative treatment for girls with nonmetastatic rhabdomyosarcoma of the genital tract: A report from the Study Committee of the International Society of Pediatric Oncology. J Clin Oncol, 17: 2117, 1999 ↩
-
Rodeberg, D. A., Paidas, C. N., Lobe, T. L. et al.: Surgical Principles for Children/Adolescents With Newly Diagnosed Rhabdomyosarcoma: A Report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. Sarcoma, 6: 111, 2002 ↩
-
Breneman, J., Meza, J., Donaldson, S. S. et al.: Local control with reduced-dose radiotherapy for low-risk rhabdomyosarcoma: a report from the Children's Oncology Group D9602 study. Int J Radiat Oncol Biol Phys, 83: 720, 2012 ↩ ↩2 ↩3 ↩4 ↩5 ↩6
-
Walterhouse, D. O., Meza, J. L., Breneman, J. C. et al.: Local control and outcome in children with localized vaginal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma committee of the Children's Oncology Group. Pediatr Blood Cancer, 57: 76, 2011 ↩
-
Puri, D. R., Wexler, L. H., Meyers, P. A. et al.: The challenging role of radiation therapy for very young children with rhabdomyosarcoma. Int J Radiat Oncol Biol Phys, 65: 1177, 2006 ↩
-
Raney, R. B., Jr., Gehan, E. A., Hays, D. M. et al.: Primary chemotherapy with or without radiation therapy and/or surgery for children with localized sarcoma of the bladder, prostate, vagina, uterus, and cervix. A comparison of the results in Intergroup Rhabdomyosarcoma Studies I and II. Cancer, 66: 2072, 1990 ↩
-
Magne, N., Haie-Meder, C.: Brachytherapy for genital-tract rhabdomyosarcomas in girls: technical aspects, reports, and perspectives. Lancet Oncol, 8: 725, 2007 ↩
-
Magne, N., Oberlin, O., Martelli, H. et al.: Vulval and vaginal rhabdomyosarcoma in children: update and reappraisal of Institut Gustave Roussy brachytherapy experience. Int J Radiat Oncol Biol Phys, 72: 878, 2008 ↩
-
Rodary, C., Gehan, E. A., Flamant, F. et al.: Prognostic factors in 951 nonmetastatic rhabdomyosarcoma in children: a report from the International Rhabdomyosarcoma Workshop. Med Pediatr Oncol, 19: 89, 1991 ↩
-
Vasquez, R., Collini, P., Meazza, C. et al.: Late relapse of embryonal rhabdomyosarcoma, botryoid variant, of the vagina. Pediatr Blood Cancer, 51: 140, 2008 ↩
-
Spunt, S. L., Sweeney, T. A., Hudson, M. M. et al.: Late effects of pelvic rhabdomyosarcoma and its treatment in female survivors. J Clin Oncol, 23: 7143, 2005 ↩
-
Ariza, M., Rafaee, T., Adeeb, N. et al.: A successful pregnancy outcome in treated vulval rhabdomyosarcoma. Med J Malaysia, 54: 371, 1999 ↩
-
Mansky, P., Arai, A., Stratton, P. et al.: Treatment late effects in long-term survivors of pediatric sarcoma. Pediatr Blood Cancer, 48: 192, 2007 ↩ ↩2
-
Kenney, L. B., Laufer, M. R., Grant, F. D. et al.: High risk of infertility and long term gonadal damage in males treated with high dose cyclophosphamide for sarcoma during childhood. Cancer, 91: 613, 2001 ↩
-
Ridola, V., Fawaz, O., Aubier, F. et al.: Testicular function of survivors of childhood cancer: a comparative study between ifosfamide- and cyclophosphamide-based regimens. Eur J Cancer, 45: 814, 2009 ↩
-
Oberlin, O., Fawaz, O., Rey, A. et al.: Long-term evaluation of Ifosfamide-related nephrotoxicity in children. J Clin Oncol, 27: 5350, 2009 ↩
-
Hishiki, T., Saito, T., Mitsunaga, T. et al.: Optimal surgical treatment and urological outcomes in boys with pelvic and urogenital rhabdomyosarcomas and soft tissue sarcomas. Pediatr Surg Int, 29: 1077, 2013 ↩
-
Raney, B., Anderson, J., Arndt, C. et al.: Primary renal sarcomas in the Intergroup Rhabdomyosarcoma Study Group (IRSG) experience, 1972-2005: A report from the Children's Oncology Group. Pediatr Blood Cancer, 51: 339, 2008 ↩ ↩2 ↩3
-
Fanous, R. N., Mayer, E. K., Vale, J. et al.: Primary renal embryonal rhabdomyosarcoma in adults: a case report and review of the literature. Case Rep Oncol Med, 2012: 460749, 2012 ↩
-
Carpentieri, D. F., Nichols, K., Chou, P. M. et al.: The expression of WT1 in the differentiation of rhabdomyosarcoma from other pediatric small round blue cell tumors. Mod Pathol, 15: 1080, 2002 ↩
-
Morotti, R. A., Nicol, K. K., Parham, D. M. et al.: An immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the Children's Oncology Group experience. Am J Surg Pathol, 30: 962, 2006 ↩
-
Walther, A., Cost, N. G., Garrison, A. P. et al.: Renal rhabdomyosarcoma in a pancake kidney. Urology, 82: 458, 2013 ↩
-
Blakely, M. L., Andrassy, R. J., Raney, R. B. et al.: Prognostic factors and surgical treatment guidelines for children with rhabdomyosarcoma of the perineum or anus: a report of Intergroup Rhabdomyosarcoma Studies I through IV, 1972 through 1997. J Pediatr Surg, 38: 347, 2003 ↩ ↩2
-
Pham, T. H., Iqbal, C. W., Zarroug, A. E. et al.: Retroperitoneal sarcomas in children: outcomes from an institution. J Pediatr Surg, 42: 829, 2007 ↩ ↩2 ↩3