INTRODUCTION
Prune Belly syndrome consists of a constellation of congenital malformations, with the three major abnormalities being laxity of the abdominal wall, bilateral undescended testes and abnormalities of the genitourinary tract. While the unusual “prune-like” appearance of the abdomen (Figure 1) is the hallmark that usually identifies these patients, their underlying renal function is the most important factor in determining their overall survival. Urinary abnormalities that have been associated with prune belly syndrome include hydronephrosis, tortuous dilated ureters, variable degrees of renal dysplasia and an enlarged bladder. The respiratory, musculoskeletal, gastrointestinal, orthopedic and cardiac systems may also be involved. There seems to be a broad spectrum of severity within this syndrome as some children with severe pulmonary and kidney disturbances will not survive the neonatal period, while others with minimal involvement will only be modestly affected.
The abdominal wall defect was first described by Frölich in 1839 and later was graphically recorded by Platt.1 Parker was the first to report a male infant with all three classic components of the prune belly syndrome.2 Osler in 1901 was the first to utilize the name “prune belly” and recognized the distended bladder associated with this syndrome.1,3 While, the title “prune belly syndrome” has persisted throughout the years, others have found this designation distasteful and utilized names such as the “triad syndrome,” “mesenchymal dysplasia” and the “Eagle-Barrett syndrome.”4-6
Prune belly syndrome affects between 1 in 30,000 to 1 in 50,000 births, with 95% occurring in male infants.7-9 A recent Kids’ Inpatient Database review revealed 133 newborn male infants with PBS of 1 420 991 births, with an incidence estimate of 3.8 cases/100 000 live births.10 Involved females present with absent abdominal musculature as well as abnormalities of the genital and urinary systems.11 There also appears to be a higher incidence in twin pregnancies as well as children born to younger mothers.8
GENETICS
No clear genetic pattern for prune belly syndrome exists, although there is a hypothesized familial inheritance pattern in some isolated cases. Prune belly syndrome been reported to occur in twins, cousins and both male and female siblings, suggesting a familial inheritance.12-23 Ramasamy et al reviewed the majority of these familial cases in 2004.12 They surmised that there may be a sex influenced, autosomal recessive inheritance pattern that may account for these isolated reports. As diseases inherited in an autosomal recessive pattern appear to occur at a higher frequency in consanguineous marriages, additional evidence for this inheritance pattern is suggested by reports by Frydman and Adeyokunnu.13,24 Frydman reported a case of prune belly syndrome in a male born to a couple in a consanguineous relationship; while Adeyokunnu reported a higher incidence of this syndrome in Nigeria, an area with known greater rates of consanguinity.13,24 Riccardi and Grum have also suggested a two-step autosomal dominant mutation with sex-limited expression that may account for the male predominance of this syndrome, as well as the infrequent number of affected family members.18 While these reports make an argument for a familial inheritance pattern, the majority of the cases appear to occur in a sporadic nature. Ives et al in their review of children affected with prune belly syndrome noted eleven pairs of twins discordant for the syndrome which speaks against an entirely genetic basis accounting for this syndrome’s occurrence.5
In addition, specific chromosomal abnormalities are also rare. Prune belly syndrome has been associated with trisomy 13, trisomy 18, trisomy 21 and Turner’s syndrome, but a specific relationship has not been demonstrated.25-27 Translocations and pericentric inversions of chromosome 9 have also been reported in fetuses that exhibit this syndrome. However, these chromosomal findings are non-specific, as not all children with this syndrome have these chromosomal abnormalities, nor do all children with pericentric inversions of chromosome 9 exhibit the prune belly syndrome.28
Associations between posterior urethral valves, Perlman syndrome, Beckwith-Wiedemann syndrome, the VACTERL association and the megacystis-microcolon-intestinal hypoperistalsis syndrome have been reported.29-32 These associations suggest a common pathogenesis between prune belly syndrome and these other conditions.
EMBRYOLOGY
There are three major theories that exist to explain the prune belly syndrome: fetal outlet obstruction, mesodermal arrest and the yolk sac theory.
Fetal Outlet Obstruction In 1903, Stumme suggested that the prune belly syndrome occurred secondary to bladder outlet obstruction.33 He surmised that this infra-vesical obstruction resulted in the bladder distension, urethral dilation and hydronephrosis characteristic of this syndrome. Dilation of the bladder was also felt to result in atrophy of the abdominal wall through venous infarction and also obstruct the normal dissent of the testicles.34 This theory was later substantiated by Gonzalez et al who recreated the phenotypic findings found in prune belly syndrome by obstructing the fetal lamb urethra at 43-45 days gestation.35 If infra-vesical obstruction were to result in the findings associated with the prune belly syndrome, it would appear that the timing and severity of this obstruction must be critical since the associated findings of this syndrome are distinct from other obstructive uropathies. Based on the development timeline of the fetus, it would seem that the obstruction would need to occur between the 13th and 15th weeks of gestation. Obstruction at this time could account for the abnormalities found in the prune belly syndrome since by this time the urachus has begun to close, significant fetal urine production has occurred and prostatic development is also just beginning.9
In humans, urethral obstruction is actually only identified in about 10-20% of cases.36 Some have postulated that the obstruction may be transient, while others have speculated that those with obstructive lesions may only represent the most severe form of the prune belly syndrome. Most of these infants that actually demonstrate urethral obstruction are from autopsy studies, therefore some feel that the urethral obstruction model may actually account for “the lethal variant” and may not account for children that survive.37 To counteract this argument, some have surmised that the hypoplastic prostatic urethra could actually function as an obstructing flap valve. In an autopsy series, Moerman et al found that the prostates of prune belly children were devoid of smooth muscle fibers and that tuboalveolar glands were reduced in number.38 They postulated that these hypoplastic prostates resulted in weakness of the prostatic walls and sacculation of the prostatic urethra. They noted this bulging was most marked along the dorsal and caudal aspects and felt that this resulted in the membranous urethra inserting more dorsally, in an oblique manner, into the prostatic urethra. It was felt that this abnormal insertion into the hypoplastic prostatic urethra could result in a flap valve mechanism, causing a functional obstruction. In this fashion, prostatic hypoplasia could be the cause and not necessarily the effect of the prune belly syndrome. Others have hypothesized that delayed canalization of the distal urethra could be the cause of obstruction.39 This delay could account for the inability to identify a true obstruction at the time of birth, as well as the development of the megalourethra that may occur in this syndrome.
The obstructive theory contends that the abdominal wall findings are secondary to pressure effects of the bladder on the developing abdominal muscles. Supporting this theory, the muscles directly over the bladder seem to be the ones that are most severely affected. In addition, Moerman et al also identified dystrophic muscle changes upon histologic analysis in their autopsy study.38 They felt that that these histologic findings could be compatible with Pinto’s description of muscular venous infarction, which he felt, was secondary to pressure from the distended urinary bladder. However, other histologic and electron microscopy studies of the abdominal wall are more consistent with developmental arrest, rather that atrophy, and fail to support outlet obstruction as the cause of the abdominal wall abnormalities.40,41
While fetal infra-vesical obstruction seems plausible, it fails to account for all of the findings associated with prune belly syndrome. Certainly there are inconsistencies with this theory and not all factors common in prune belly children can be explained by this model.
Theory of Mesodermal Arrest This theory suggests that the etiology of the prune belly syndrome is secondary to a noxious insult to the embryonic mesoderm. During normal fetal development, the mesoderm flows out between the ectoderm and endoderm. The mesoderm then divides into segments medially and laterally, which form the somites and the lateral plate, respectively. The lateral plate then further splits into a visceral and somatic layer. The somatic layer between the somites and the lateral plate forms the nephrogenic mesoderm, which is the precursor of the mesonephros, wolffian duct and metanephros. The somatic layer of the lateral plate mesoderm is the source of abdominal wall musculature, while the visceral layer of the lateral plate mesoderm gives rise to the smooth muscle of the urinary and gastrointestinal tracts.9 Many believe that abnormalities of the mesoderm could account for the syndrome’s abdominal wall and genitourinary findings.
Stephens and Gupta proposed that abnormalities of the urinary tract could be explained by abnormal development of the mesonephros between the 6th and 10th weeks. Normally, the Wolffian duct, which is a derivative of the mesoderm, terminates caudally into the urogenital sinus and becomes incorporated into the future membranous urethra and demarcates the verumontanum. The wolffian ducts and ureteric buds are further incorporated into the trigone, with the ureteric buds growing into the nephrogenic blastema. Abnormal incorporation of the terminal wolffian ducts could produce prostatic hypoplasia, dilation of the prostatic urethra and even valve-like obstructions. In addition, involvement of the ureteric buds could lead to abnormal ureteral development and even dysplastic renal development if the interaction with the nephrogenic blastema is abnormal.42 Simultaneous failure of the somatic layer of the lateral plate mesoderm could then account for the abdominal wall defects.
Ives proposed that the mesodermal defect could occur as early as the 3rd week of gestation. She felt that at the division of the embryonic disc, which normally occurs at the 3rd week, could account for the discordant occurrence seen with monozygotic twins.5 Others postulate that the mesodermal abnormalities could also account for difficulties in testicular descent. They feel that abnormalities in the testicular gubernaculums, also derivatives of the mesoderm, could result in unresponsiveness of these tissues to hormonal influences and thus not allow the testes to descend normally.43-46
However, even the mesodermal arrest theory fails to explain all of the factors associated with the prune belly syndrome. Further evidence and explanation is necessary to fully support this theory as the cause of this syndrome.
The Yolk Sac Theory Stephens has proposed an additional theory to explain the features associated with the prune belly syndrome. He hypothesizes that in children with prune belly syndrome, the bladder and posterior urethra are derived from a larger portion of the allantois than is normally accepted and that an abnormal amount of the yolk sac is retained as well. This abnormal amount of yolk sac could affect the abdominal wall development and result in the “prune-like” appearance. In addition, the allantoic diverticulum, may be incorporated into the urinary tract and account for the enlarged urachus, bladder and prostatic urethra apparent in the prune belly syndrome.42 However, this theory fails to account for all of the abnormalities seen in the renal and genital tract.
CLINICAL FEATURES
Genitourinary Abnormalities
Kidneys There is a wide variation in renal presentation, ranging from normal to severely dysplastic, nonfunctional kidneys.47 It is the degree of renal dysplasia and the child’s overall renal function that usually determines their prognosis. These renal issues and its resultant pulmonary hypoplasia account for a 20% chance of stillbirth or death in the neonatal period, with an additional 30% progressing to renal failure in the first two years of life.33,48 Histologically, the most dysplastic kidneys are similar to those described by Potter demonstrating few nephrons and other disorganized features.9
Hydronephrosis is also a common finding (Figure 2A, B) and currently is usually detected on prenatal ultrasounds.49 One must remember that the degree of hydronephrosis does not necessarily correlate with renal function, the degree of ureteral dilation or the abdominal wall laxity.50 In addition, it is not unusual to identify asymmetric renal involvement, with one kidney demonstrating severe hydronephrosis and/or dysplasia as compared to the normal contralateral side.51 Ureteropelvic junction obstruction has also been reported, however non-obstructive hydronephrosis is the general rule.52 In those patients with preserved renal function, it appears that urinary tract infections may pose the greatest risk to damaging their kidneys.53
Ureters The ureters are elongated, dilated and tortuous (Figure 3), with the lower one third being more profoundly affected as compared to the proximal portion.33 Vesicoureteral reflux occurs in up to 75% of patients and the ureteric orifices may be wide and patulous.9 Fluoroscopic images of the ureters may demonstrate ineffective peristalsis, however typically they are unobstructed and drain adequately.33 Although rare, there are reports of obstruction at both the ureteropelvic junction and ureterovesical junction.38,43,54
Histologic examination of the ureters reveals alterations in the ureteral wall architecture. Hypertrophy and smooth muscle hyperplasia, which may be seen in patients with posterior urethral valves and vesicoureteral reflux, is typically absent in the prune belly syndrome. Instead, smooth muscle is sparse or even absent and may be replaced with cellular fibrous tissue.4,38 There also appears to be an increase in the ratio of collagen to smooth muscle in the ureteric muscular layer, especially if vesico-ureteral reflux is present.55 Although present throughout the ureter, the proximal portion of the ureter appears to have more smooth muscle cells as compared to the distal portion.4,56 There also appears to be a decrease in the number of nerve plexuses and Schwann fibers within the ureter as well. These histologic features may interfere with cell-to-cell impulse propagation and result in poor ureteral peristalsis. These features should also be taken into consideration if ureteral surgery is undertaken in these patients.
Bladder The bladder in patients with the prune belly syndrome is typically enlarged and thickened (Figure 4), but trabeculations are usually absent.9 This is in sharp contrast to the bladder of a patient with posterior urethral valves, which is usually markedly trabeculated. During a cystogram, the bladder therefore is typically smooth walled and may demonstrate an urachal remnant or diverticulum. Urachal remnants may attenuate the bladder to the abdominal wall. Urachal cysts or a fully patent urachus are present in 25-50% of the cases.36,42,43 The trigone is typically enlarged with laterally placed, patulous ureteral orifices, which accounts for the common association with vesicoureteral reflux.9 The trigone also typically funnels into a poorly defined, wide open bladder neck.39
Histologically, the bladder appears to demonstrate two patterns in patients with prune belly syndrome. In patients with definite lower urinary tract obstruction the thickened bladders demonstrate an increased muscle quantity and a normal collagen to muscle ratio. Bladders without obstruction demonstrate a thinner bladder wall with an increase in the collagen to muscle ratio.57 The bladder innervation though appears to be normal with a normal distribution of ganglion cells.4
Urodynamic evaluation of the prune belly bladder typically reveals a large capacity bladder with normal resting pressures.4 These patients usually also demonstrate a delayed first sensation of fullness with poor bladder contractility.58,59 Bladder instability with uninhibited detrusor contractions may occur, but usually are rare.58-60 Kinahan et al reported in their series that 44% of their patients were able to void spontaneously, with normal voiding pressures and low residual volumes; while the others required clean intermittent catheterization.59 Certainly this poor contractility as well as the massive vesicoureteral reflux seen in these patients would account for the high levels of residual urine that may be seen in these individuals.
Prostate and Posterior Urethra The prostatic urethra is typically elongated and tapers at the membranous urethra (Figure 5). In addition the bladder neck is usually widely patent and dilated. These characteristics give the prostatic urethra a typical “triangular shape” on radiographs.33 The anterior prostatic wall is typically shorter than the posterior wall. The verumontanum is usually small or absent and a utricular diverticulum may be present.33 A true obstruction at the level of the prostatic or membranous urethra is rare, only occurring in 10-20% of cases.33 As discussed earlier, these patients who demonstrate true urethral obstructions are generally considered to be the most severe form of prune belly syndrome. Without a true urethral obstruction, Stephens has postulated that there may be kinking of the prostatic urethra secondary to the differences in length between the anterior and posterior prostate. This kinking may then account for a functional obstruction by creating a flap valve of the urethra.42
Histologic analysis of the prostate in prune belly patients has revealed severe hypoplasia, with minimal smooth muscle and tubuloalveolar glands.38 In comparison, the prostates from patients with posterior urethral valves revealed a normal amount of smooth muscle and glandular parenchyma that was compressed but otherwise normal. This lead Moerman to speculate that the dilated, hypoplastic prostate is the cause, rather than the effect, of the prune belly syndrome.38
Anterior Urethra Typically the anterior urethra in these patients is normal, although there have been reports of both urethral atresia and microurethras, as well as the development of the megalourethra in these patients. Those patients with either urethral atresia or a microurethra typically only survive secondary to a patent urachus. Some have suggested that these narrow urethras are normally developed, but underused and may therefore be managed with progressive dilation.61
The two types of megalourethra, scaphoid and fusiform, may be associated with this syndrome. In the fusiform variety, the corpora cavernosa is deficient as well as the spongiosum. This form is more severe and may be associated with renal dysplasia and other lethal abnormalities.62 Scaphoid megalourethra, which is the more common and less severe form, is characterized by only a deficiency of the spongiosum, with preservation of the corpora cavernosa, glans and fossa navicularis.33 It is believed that a mesodermal defect could account for these urethral maldevelopments.63
Accessory Male Sex Organs Abnormalities of the vas deferens, epididymis and seminal vesicles have been noted. Given the common mesodermal derivation of these organs, these findings are not surprising. The epididymis is commonly detached from the testes and the vas has been found to drain ectopically.64 In addition, the seminal vesicles may be dilated or even atretic or absent.42 All of these findings, along with the abnormal testicular histology are significant in regards to these patients’ future fertility.
Testis Bilateral cryptorchidism is considered a hallmark of this syndrome, with the majority of these testes being intra-abdominal above the level of the iliac vessels.33 The gubernaculums originally were suggested to be normal, although elongated and attached to the internal inguinal ring; however Elder et al have suggested that the gubernaculum may actually be abnormal and possibly even absent.4,45 This explanation may also account for the maldescent of these testes, in addition to the theory of obstruction from the distended bladder.
Histologically these testes also demonstrate a variety of abnormalities. Orvis et al have reported a reduction in the number of spermatogonia, as well as Leydig cell hyperplasia.65 In addition, Massad et al have demonstrated atypical germ cells with abnormal nuclei.66 These abnormal germ cells resemble intratubular testicular neoplasms and thus children with prune belly syndrome must be closely monitored for the development of testicular tumors. Indeed, there have been documented cases of testicular tumors developing in these patients, but overall the risk does not appear to be greater than other patients with undescended testes.67-69
Sexual Function and Fertility Sexual function in patients with prune belly syndrome appears to be intact. Provided that the corpora cavernosum are normal, these patients may experience normal erections and orgasm and may maintain satisfying sexual relationships.70 However, given the open bladder neck, these patients experience retrograde ejaculation and tend to be azoospermic.70,71 There are no reports of fathers with prune belly syndrome reproducing naturally. To date the only reported case of paternity in a patient with prune belly syndrome involved intracytoplasmic sperm injection.72 Clearly, the infertility associated with this syndrome is multifactorial as there are histologic abnormalities of the testes and prostate, as well as anatomic abnormalities of the prostate, bladder neck, vas deferens, seminal vesicles and testicles.
Extragenitourinary Abnormalities
Abdominal Wall The characteristic wrinkled, “prune-like” skin of the newborn is one characteristic finding in children with prune belly syndrome (Figure 1). This appearance is secondary to skeletal muscle hypoplasia that occurs in all three muscle layers of the abdominal wall.40 The upper rectus muscles and outer oblique muscles are usually better developed and the degree of hypoplasia may be variable and asymmetric 9, 33. The order of involvement from most to least involved is: transversus abdominis, rectus abdominis below the umbilicus, internal oblique, external oblique and rectus abdominis above the umbilicus.6 The more developed upper rectus abdominis muscle results in cephalic displacement of the umbilicus. Indeed, the diminished muscle mass may allow one to see the bowel peristalsis through the skin.
The impaired abdominal wall may hinder a newborn’s ability to sit from the supine position and may delay the child’s ability to walk.33 Otherwise, there appears to be no other hindrance to normal physical activity. The lack of abdominal muscle may also result in a poor coughing ability and may contribute to an increased incidence of respiratory infections.73 Others surmise that the deficient abdominal muscles, may also contribute to constipation.39 Overall however, it appears that although the abdominal wall laxity may be visibly profound, the physical limitations from this deficiency are actually quite minimal.
Evaluation of the abdominal wall has revealed normal nerve distribution and blood supply to the muscles.40,74 The muscle fibers occur haphazardly and in the most severe cases are entirely absent. Microscopically, the fibers may be replaced by a thick collagenous layer.9 In addition, Mininberg et al utilized electron microscopy to visualize Z-band disorganization and glycogen aggregates.40
Orthopedic Abnormalities Orthopedic abnormalities occur in 45-63% of prune belly patients.75 The most frequent anomalies are hip dislocation and talipes equinovarus.51 More severe deformities such as limb deformities and arthrogryposis appear to be secondary to oligohydroamnios causing compression of the developing fetus.51,76 In addition pectus carinatum and pectus excavatum deformities are commonly present, while scoliosis is the most common spinal abnormality.76,77 Growth has been found to be impaired in 32% of prune belly patients and has correlated with poor renal function.78 The growth retardation appears to be most profound in the first year of life, but catch-up growth appears to remain incomplete throughout life.78
Gastrointestinal Abnormalities Gastrointestinal anomalies appear to occur in up to 30% of affected prune belly children. Abnormalities such as malrotation, atresia, stenosis and volvulus appear to result secondary to persistence of the embryonic mesentery.79 The same defects in suspension may also allow the spleen to wander freely and splenic torsion has been reported.80 Gastroschisis, imperforate anus and omphaloceles have all been reported to occur in prune belly patients.79,81-83 Constipation may also occur and may be the result of absent abdominal muscular tone. In addition, Hirshsprung’s disease has been reported to occur in these patients and may also contribute to the constipation. Because of these reported gastrointestinal associations, it has been recommended that all prune belly patients undergo radiographic evaluation of their GI tract.79
Cardiac Abnormalities Cardiac abnormalities have been reported to occur in 10% of prune belly patients.51 Associated cardiac findings include ventricular and atrial septal defects, patent ductus arteriosus and tetralogy of Fallot.51 Given these associations, it is recommended that all prune belly patients undergo cardiovascular evaluation.
Pulmonary Abnormalities As pulmonary development in utero is dependent on adequate amounts of amniotic fluid, fetuses with renal failure and severe renal insufficiency may also develop pulmonary hypoplasia. In the worst cases, severe pulmonary hypoplasia is incompatible with life and may result in death in the perinatal period.51 It appears that 20% of newborns will die in the perinatal period secondary to pulmonary issues.71,84 Of those that survive, 55% of patients will suffer prolonged, significant pulmonary problems.78 The lax abdominal wall may impair mechanical ventilation and can increase the incidence of pneumonia.85,86 These same issues may also contribute to the development of bronchitis and respiratory compromise that may occur after anesthesia and respiratory tract infections.78,86
There are also a subset of patients with incomplete features of this syndrome and have been dubbed the “pseudoprune”. Often these individuals will not have the characteristic abdominal wall findings, but will suffer from bilateral cryptorchidism and also demonstrate the associated urinary tract findings.87 These patients are still at risk for developing renal failure and therefore must be followed closely.87
PBS in Girls Five percent of prune belly children are female. Associated abnormalities in these children include the typical abdominal wall appearance and urinary tract abnormalities (Figure 6).11 Additional findings in these females included a high rate of vaginal atresia, uterine duplication and imperforate anus. Reinberg et al, in their series of females with prune belly syndrome also demonstrated a high rate of renal hypoplasia and dysplasia, similar to their male counterparts.11 Urethral atresia, uterine duplication, and anorectal anomalies occurred frequently in these girls. The perinatal mortality rate was high; of the four surviving patients, renal failure developed in two and renal transplantation was required.11
PRESENTATION AND EVALUATION
Prenatal Diagnosis and Intervention
The diagnosis of prune belly syndrome may be suspected as early as 11 weeks of gestation via prenatal ultrasound.88 Associated sonographic findings include oligohydroamnios or anhydramnios, hydroureter, a distended bladder and a thin, attenuated abdominal wall.89 Megacystis megaureter syndrome or posterior urethral valves, may have similar findings in fetal ultrasonogram. Although modern ultrasonic capabilities and visualization have improved, it can still be difficult to establish a definite prenatal diagnosis. Previous reports have shown that prenatal diagnostic accuracy to ascertain the etiology of hydronephrosis varies between 30-85%.90
The identification of the above prenatal findings have prompted some to either recommend in-utero placement of a vesicoamniotic shunt in order to decompress the urinary tract and establish a normal amount of amniotic fluid or suggest termination of pregnancy.91-94 The degree of collecting system dilatation, however, does not represent the severity of obstruction nor does it reflect the post-natal renal function. Thus it may be difficult to justify the need for any intervention or termination using degree of hydronephrosis alone.95 Criteria used for in-utero shunting include a normal karyotype, severe oligohydroamnios and normal fetal renal function.94 Decision to intervene is based on serial evaluations of fetal urine chemistries such as sodium, chloride, osmolarity, microglobulin, and total protein.96 Fetuses with high urine sodium and osmolarity values have the poorest prognosis. In the rare instance of progressive oligohydramnios with urethral atresia97 or potential obstruction during labor due to massively distended bladder intervention may be beneficial.91 Despite several case reports documenting the ability to perform this procedure, reviews have failed to demonstrate a sustainable improvement in renal and pulmonary function.90,98,99 In addition, complications of shunt placement have occurred including traumatic gastroschisis secondary to an abdominal wall defect from a misplaced shunt.100 Freedman et al also demonstrated a 38% mortality rate and 29% renal failure rate in children that underwent intrauterine interventions.97 It would seem that in-utero intervention has certain potential, but does not yet achieve the desired renal or pulmonary improvement benefit. Given these circumstances, fetal intervention requires a careful risk/benefit assessment and a thorough discussion with the child’s parents.
Peripartum Care Since the newborn may require ventillatory support and advanced neonatal care, it is prudent to have the delivery where neonatal care facilities are available. Delivery may be complicated by preterm labor, chorioamnionitis or fetal entrapment due to the enlarged bladder. The associated pulmonary hypoplasia in infants with prune belly syndrome can lead to pneumothoraces. For neonates who have pulmonary hypoplasia, advanced support such as high-frequency ventilation and possibly inhaled nitric oxide (iNO) will be required.101 An experienced obstetric and neonatal team should be present at delivery.
Neonatal Period The characteristic prune-like appearance of the abdomen usually suggests the diagnosis (Figure 1). A multi-disciplinary approach is required for the initial evaluation, specifically involvement of the neonatologist, nephrologist and urologist. Involvement of other disciplines will depend on the clinical profile. If there is cardiac or pulmonary involvement, evaluation and treatment of these take precedence during the immediate post natal examination. Genitourinary involvement may be assessed once the child is stable.
Evaluation of Renal Function An initial creatinine serves as a baseline but the value may be related to maternal creatinine. Serial serum creatinine measurements are essential to determine the neonatal renal function as well as to monitor the trend. A rise in creatinine over the first several weeks portends a poor prognosis. If the creatinine is noted to nadir at less than 0.7 ng/dl, it has been noted that subsequent renal failure is less likely.102 In addition, serum electrolytes should also be monitored and may be helpful in assessing overall renal function.
Imaging Radiographic evaluation of the newborn should begin with chest radiography. This will help rule out any associated pulmonary complications as a result of oligohydramnios which can be seen with PBS, specifically pneumothorax, pneumomediastinum, and pulmonary hypoplasia.103 Renal and bladder ultrasound can assess parenchyma, the presences of cortical cysts and bladder distension. A voiding cystourethrogram (VCUG) should be obtained (Figures 3, 5) in the presence of renal insufficiency104 to differentiate between obstruction and stasis. Ideally, prophylactic antibiotics should be started before a VCUG is obtained to minimize the risk of infection. In the newborn with normal renal function and clinical evidence of bladder emptying, a VCUG can be delayed, since nearly 70% of children have vesicoureteral reflux. 105, 106 Nuclear medicine renal scan with 99mTcDMSA or 99mTcMAG3 helps to identify functioning renal tissue. However, stasis due to ureteral dilation and tortuosity may preclude any diagnosis of obstruction with lasix washout renography (Figure 7). Renography can be obtained after 4 weeks to allow for changes during neonatal physiology.
MANAGEMENT
Spectrum of PBS
Since there is a wide spectrum of presenting characteristics in patients with PBS three major categories of presentation in the neonatal period was described by Woodard.107 Though there is no clear delineation among the categories, this classification helps planning the evaluation and management of the neonate.
Category 1 The renal dysplasia or urethral obstruction secondary to atresia results in marked oligohydramnios. This in turn results in severe pulmonary hypoplasia. A constellation of other system involvement may be present. The pulmonary hypoplasia or renal failure commonly results in death in the neonatal period. Since these neonates have other life threatening issues, only limited urological intervention is indicated, often it is limited to bladder catheterization.
Category 2 They have varying degree of renal insufficiency, upper tract dilatation and vesicoureteral reflux. As they seldom have life threatening early neonatal pulmonary or renal insufficiency, the evaluation and management of the urinary tract assumes importance. They require an individualized approach due to varying degrees of presentation. Specific treatment of the dilated urinary tract in this group has remained controversial for the past 25 years.108, 109 There is an overlap between the clinical features of Category II and Category III patients and differing proportions of these are included in reported series. In addition, the long term follow up is limited. Hence it is difficult to compare the results of various series that advocate either aggressive surgical management or a conservative approach. Patients in category 2 require an individualized approach due to varying degrees of presentation. Since infection and progressive renal insufficiency is the major cause of morbidity and mortality in this group, aggressive surgical therapies were performed and as a result, anatomical and functional improvement was noted.84,108,110 Improved renal outcome and decrease in urinary tract infections was documented with ureteral tailoring, shortening and reimplantation, thus minimizing stasis and reflux.84,110 Others have advocated close observation of these patients with monitoring for urinary tract infection and surgical intervention for those with urinary tract obstruction or ongoing bacterial infection.109 Woodhouse et al, in a series of 47 cases, had 13 neonates who presented with infection and gross urinary tract dilatation. High diversion was carried out in twelve. Further upper tract reconstruction was attempted in six of these but normal micturition and stable renal function were achieved only in three.111 However, due to the wide variation of presenting characteristics in this patient population and the limited published data, no outright recommendation can be made regarding patients who fall into this category. The treatment plan should be tailor made depending upon the co-morbidities, renal function, infections, overall clinical picture and the preferences of the family.112
Category 3 This group comprises children with incomplete or a mild triad of features, mild to moderate uropathy, no renal dysplasia, stable renal function and no pulmonary hypoplasia. Their renal function is usually preserved. They require regular monitoring with ultrasound and creatinine. Unless there is worsening of the renal function or recurrent UTI, they do not require any surgery of the urinary tract. In the absence of indication for urinary tract reconstruction, bilateral orchiopexy is carried out in infancy. Any deterioration in renal function or new urinary tract infection requires further evaluation. Urodynamics may be required to assess the bladder and adequacy of voiding.58
Surgical Treatment
Urinary Diversion Urinary diversion may be required during initial management, in view of urosepsis, urinary tract obstruction or worsening renal function. If voiding is inefficient because of urethral stricture, valves or atresia, urinary diversion will be required. In neonates with bladder outflow obstruction, the urachus is patent but since it closes in the first few weeks of life, drainage through urachus is unpredictable.
Vesicostomy When indicated, cutaneous vesicostomy is a quick and efficient method to divert the urine at the level of the bladder and Blocksom’s technique is commonly employed. It is important to construct the stoma using the dome of the bladder. Since the bladder is large and floppy, if the cystotomy is made lower down in the anterior bladder wall it is prone to prolapse and obstruction. A larger stoma is recommended in PBS since stomal stenosis is common. A patent urachus or a large diverticulum at the urachus are encountered frequently. If present, the diverticulum or patent urachus may be excised at the time of vesicostomy.
Supravesical Diversion In the majority of cases, vesicostomy is adequate for effective decompression of the urinary system. In the setting of massive reflux, a single loop ureterostomy may allow for adequate decompression of both kidneys while simultaneously allowing for normal bladder cycling. However, in the setting of obstruction at the ureterovesical or ureteropelvic junction, proximal urinary diversion is recommended. If ureterovesical reconstruction is anticipated, it is preferable to avoid a high loop ureterostomy, since the upper ureter is utilized for the reconstruction and the vascularity of this segment may be compromised by the loop ureterostomy.56,113 A low loop ureterostomy may adequately decompress both the ipsilateral kidney as well as the bladder without endangering later reconstruction. End cutaneous ureterostomy may be considered if upper ureteric obstruction is not suspected. In addition to relieving obstruction, the upper ureteric anatomy is not disturbed and there is a decrease in the caliber of the severely dilated ureter, which is desirable for future reconstruction.113 Another alternative for upper tract drainage is cutaneous pyelostomy. Infants requiring temporary diversion requiring a complete reconstruction planned at a later date may benefit from initial pyelostomy followed by bilateral tapered ureteroneocystostomy, reduction cystoplasty, bilateral orchiopexy and abdominoplasty once they are optimized.114 Since “total reconstruction” however has little role in the modern management of prune belly syndrome, rarely will pyelostomies be indicated. Temporary percutaneous nephrostomy drainage is a safer option for temporary diversion in an infant who is acidotic and septic, unfit for anesthesia.
Reconstruction
Reconstruction pertinent to urogenital system may be classified as orchidopexy, abdominoplasty and urinary tract reconstruction. Undescended testes are a hallmark of Prune Belly Syndrome and there is universal agreement regarding the need for orchidopexy. The abdominal wall defect may range from mild divergence of the rectus muscles to severe deficiency of the abdominal wall and decision for abdominoplasty depends largely on the extent of deformity and desired cosmesis. Recently, improvement in bladder emptying is an additional consideration to offer abdominoplasty in full blown PBS.115 True obstructive uropathy, recurrent urinary tract infections and chronic renal insufficiency in anticipation of renal replacement therapy are relative indications for surgical intervention. It is unclear if correction of high grade reflux and/or restoration of normal caliber and capacity of urinary tract have any effect on minimizing renal deterioration or reducing the risk for infections and there is considerable controversy regarding the indications and timing of urinary tract reconstruction. In order to improve detrusor function, reduction cystoplasty was suggested116 but it did not seem to decrease bladder capacity or improve voiding dynamics in the long-term.117 As there are no definite criteria to assign the category and classification and as the natural history is variable, it is difficult to compare the outcomes of different series.
Urethra The spectrum of urethral abnormalities includes urethral atresia or microurethra, megalourethra or obstruction at the prostato-membranous urethra. Successive urethral dilation as described by Passerini-Glazel et al using the P.A.D.U.A. technique has been suggested for urethral atresia but the results have not been uniformly successful.61 If vesicostomy is present, a through and through dilatation may be possible. Four out of six boys reported by Gonzalez et al required some form of supravesical diversion after multiple unsuccessful attempts at dilatation.118 Initial perineal flap urethrostomy followed by reconstruction may be required.119
As previously mentioned, megalourethra may be either the fusiform or the scaphoid type. The scaphoid megalourethra is more common. The technique was initially described by Nesbit in 1955.After a circumcising incision and degloving of the penis he performed a longitudinal reduction urethroplasty is performed over a catheter. This technique is straightforward, avoids overlapping suture lines, and its principles remain in wide use today.120 An alternate technique is urethral placation, as described by Heaton and colleagues.121 Reconstruction may be technically challenging in the fusiform variant, since there is no surrounding spongiosum and the urethra is often massively dilated. The options are limited, due to lack of periurethral tissue. The phallus is usually greatly enlarged and can be more capacious than the native bladder. Nevertheless, there have been reports of phallic reconstruction in these patients with satisfactory cosmetic and functional outcomes.122 The rarity of the defect precludes any generalizations with regard to surgical management; each case must be considered individually.120
Since the prostatic urethra is hypoplastic, there is an abrupt change in caliber at the prostatomembranous junction but true obstruction is rare. Occasionally prostato-membranous urethral obstruction, classified by Stephens as a type IV valve, is present. It is caused by redundant tissue in the membranous urethra and can be incised transurethrally.123 Woodhouse et al followed up 29 patients in Category III who were well at birth, with stable renal function. When there was upper deterioration, the obstruction was noted to be generally in the urethra, which was relieved endoscopically either by resection of a narrow ring or by full length urethrotomy.111 In children with PBS with high post-void residuals, increasing hydroureteronephrosis or VUR, internal urethrotomy may be considered though no long term success has been demonstrated.124
Bladder Urodynamic evaluation of the bladder is indicated in the setting of urinary tract infections and large residual volumes.58 Evidence of resistance at the vesicourethral junction has been postulated to contribute to the large post void residuals along with poor bladder contractility and high grade vesicoureteral reflux. In children who are unable to maintain low residual volumes some authors have proposed internal urethrotomy to lower the sphincter resistance and allow for improved emptying of the bladder. 58, 109 Additionally, clean intermittent catheterizations can facilitate improved bladder emptying.59
Reduction Cystoplasty Poor bladder function along with large capacity and massive vesicoureteral reflux can lead to decreased bladder emptying. In order to enhance bladder emptying, Perlmutter initially proposed reduction cystoplasty as a means to reconstruct the shape of the bladder to a more spherical appearance.116 However the bladder volume returned to baseline or increased when compared to pre-reduction cystoplasty volumes in long term follow up.117 No difference is noted on urodynamics to patients with reconstruction versus those without.59 Hence reduction cystoplasty is reserved for removal of a dilated urachal segment which may be seen with urethral abnormalities, or to excise a large urachal diverticulum.
Upper Urinary Tract
Ureteral Reconstruction Vesicoureteral reflux occurs in approximately 70% of PBS children. Conservative approach in these patients consists of antibiotic prophylaxis and serial monitoring of renal function with goals to maintain renal function and avoiding any ureteral surgery.113 Initial proximal diversion with later reconstruction was noted to yield results that are equally successful and comparable with primary reconstructive surgery carried out in early infancy or later childhood.115 In the setting of recurrent pyelonephritis or progressive renal decline, ureteric tailoring, straightening and reimplantation after excising the redundant dilated lower ureter, is recommended. Ureteral tapering and reimplantation can be performed using plications or an excisional with equal success.106 Submucosal tunneling may be challenging if plications are performed due to the bulk of the resultant ureter. Occasionally the redundant tortuous distal ureter can be excised with standard reimplantation of a more normal sized proximal ureter.
Orchidopexy Undescended testicles are one of the hallmarks of this syndrome. The timing of orchidopexy depends on fitness for anesthesia and need for concomitant abdominoplasty or urinary tract reconstruction. Since the endocrine function is preserved and there is a malignant potential, earlier orchidopexy is desirable. Most of these testes are intra-abdominal, high in the posterior pelvic wall overlying the iliac vessels, and hence spontaneous descent is unlikely. Standard orchidopexy, without division of the testicular vessels may be more successful if the orchidopexy is done during early infancy.125 Since the testes are located in the abdomen at the level of the iliac vessels, adequate proximal mobilization is difficult with the standard inguinal approach and trans-abdominal approach is preferred. If concomitant urinary reconstruction or abdominoplasty is being performed, the vessels can be mobilized till the origin to ensure adequate mobilization for single stage orchidopexy. The primary objective of orchiopexy is to preserve the hormonal function and to enable easy examination, since malignant potential has been documented.66 In addition, though the fertility potential is compromised, assisted reproductive techniques may be possible. There is a single report of mature spermatozoa in the post-coital urine in a patient who underwent neonatal orchidopexy.115 Testosterone levels were found to be higher in boys who underwent orchidopexy in early infancy compared to those who had staged or Fowler-Stephens orchidopexy at an older age. Hence orchidopexy as early as six months has been recommended.115 If no additional urological procedure is indicated and abdominoplasty is not planned, laparoscopic orchidopexy is an option to be considered. Dissection may be carried out till the origin of the testicular vessels, thus facilitating single stage orchidopexy. The altered anatomy of these children presents an unusual landscape to the laparoscopic procedure. Since the ureter and the kidneys are often grossly dilated, care must be exercised during dissection.126 Laparoscopic Fowler-Stephen’s technique is executed either as a one stage or staged approach with reasonable success when a single stage orchidopexy is not possible.126,127 Microvascular autotransplantation is an alternative for high intra-abdominal testes that cannot be brought down to the scrotum despite adequate mobilization.128
Long-term Follow-up of Orchidopexy Normal erections and orgasm was experienced by 9 patients between 16 and 28 years of age who underwent orchidopexies in late childhood or adolescence. Seven of these had retrograde ejaculation. Two of these were anorchic and of the remaining seven, six had normal testosterone levels. Though infertile, these men were sexually active.70 Orchidopexy can be performed using either an open or laparoscopic approach if completed within the first few years of life. Often, the testicles can be brought down to the scrotum without a Fowler-Stephen’s approach.110 Patil and colleagues reviewed the outcome of orchidopexy in 41 boys 20 underwent bilateral 1-stage orchidopexy, 10 with unilateral 1-stage and contralateral 2-stage orchidopexy, and 11 underwent. Except for two who underwent bilateral single stage orchidopexy, others had satisfactory outcome sufficient to induce puberty and maintain sexual function. Division of gonadal vessels did not seem to adversely affect the outcome.127 In a series of 32 boys with PBS who underwent bilateral orchidopexy, 26 did not require ligation of testicular arteries. Unilateral and bilateral ligation of pedicles was performed in 2 and 4, respectively. Five testes had atrophied, for which three underwent ligation of the artery.112
Abdominal Wall Reconstruction Similar to the urinary tract, the spectrum of abdominal wall defect extends from mild protuberance to severe laxity and bulge. In those with minimal deficiency, there may be improvement as they grow up and no intervention may be required. However, most children with PBS of Category II have significant abdominal wall defect. The abdominal wall defect may not have life threatening consequences but the inefficient contraction of the abdominal wall muscles is believed to affect bladder, bowel, and pulmonary function.129 Consequently, abdominal wall repair might improve coughing and defecation, though there is no evidence for the same.115 If concomitant urinary tract reconstruction is anticipated, it is prudent to wait till a clear picture of the urinary tract and voiding function is obtained and the reconstruction is planned. If no urinary tract reconstruction is anticipated, it may be combined with orchiopexy during infancy. Abdominoplasty along with orchidopexy at 6 months enables adequate mobilization of the testes. Since these infants have underlying pulmonary hypoplasia101 and the breathing may be further compromised by abdominoplasty, the benefit in terms of successful orchidopexy and reconstruction should be weighed against the anesthetic risk, since mortality due to pulmonary issues have been documented following neonatal reconstruction.115
Randolph Technique Randolph et al 130 initially described abdominal wall reconstruction based on electromyography data. EMG mapping indicated that the most severely affected area is the infraumbilical region. Hence they recommended a transverse incision from the tip of the 12th rib over the anterior superior iliac spine down to the pubic symphysis and back up to the contralateral anterior superior iliac spine and tip of the 12th rib. A parallel incision is made with the excess skin pulled taut. Full thickness removal of the skin, underlying muscle and peritoneum was performed. Full thickness reapproximation of the healthy inferior margin of the fascia, including the periosteum is performed at the anterior superior iliac spines and pubic symphysis using non-absorbable suture. The fascia is closed using interrupted suture followed b skin reapproximation. One series of 16 patients noted in 7 patients laterally bulging of the abdominal wall.106
Ehrlich Technique Ehrlich first described his methodology in 1986 and later modified it in 1993.131, 132 The technique involves a vertical incision with preservation of the umbilicus. There is extensive mobilization of the lateral muscular layer from the overlying skin and subcutaneous tissue, followed by a vest over pants closure. This closure brings the unaffected lateral musculofascial layer into the midline and allows for removal of excess skin and preservation of the umbilicus.
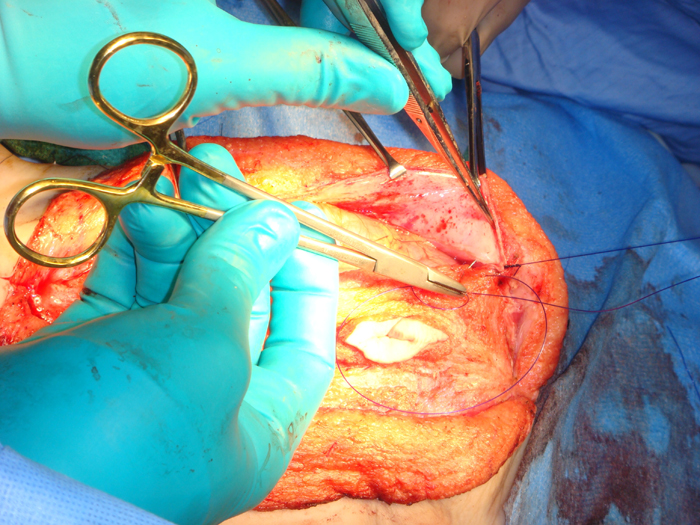
Monfort Technique The Monfort technique (Figure 8) also utilizes a vertical incision.133 Using an elliptical incision from the xiphoid to the pubic symphsis and a second incision around the umbilicus for preservation, the underlying subcutaneous layers are dissected off the fasica laterally to the anterior axillary line. A vertical incision is made lateral to the superficial epigastric arteries. The posterior parietal peritoneum is incised. The central island muscle flap is secured to the posterior parietal peritoneum. The mobilized lateral muscle flaps are brought over the central muscle flap and reapproximated in midline, similar to a vest over pants technique. This technique increases the anterior abdominal wall thickness, creates a waistline and reduces any bulge. Other authors have reported satisfactory results with good patient satisfaction using this technique. 134 Since the periumbilical skin in the midline is not excised in the Montfort technique, the umbilicus often appears too high after the repair. A neo-umbilical reconstruction along with Montfort abdominoplasty has been suggested.135 An almond shaped skin island is preserved at the level of the iliac crest in the midline, attached to the dermis. This is invaginated by placing absorbable sutures from underneath the fascia. Another modification to this technique has been described by Furness et al.136 In this approach, the peritoneal cavity is not entered and the abdominal wall dissection is minimal. If orchidopexy has been performed earlier, this technique is useful to avoid entry into the peritoneal cavity.
Firlit Technique In this technique, the laxity of the abdominal wall is assessed by grasping the midline with towel clips and lifting the abdominal wall till it is taut. An elliptical skin incision is then made around the base of the redundant skin from the xyphoid to the pubis preserving an island of the umbilical skin. The excess skin is excised and the fascial plate laxity is corrected with a plicating technique with full thickness vertical folds. The peritoneal cavity is note entered. Hence this procedure is an option if no concomitant intraperitoneal procedures are required. Authors have reported satisfactory outcome in 13 patients who underwent the procedure.136
Laparoscopic Assisted Abdominal Wall Reconstruction Laparoscopic-assisted repair has been described to minimize violation of the peritoneal cavity.137 Peritoneal cavity was visualized using a telescope introduced through a 5 mm sub-xiphoid trocar, while vertical abdominal wall plication of the fascia was done by Firlit technique.136 In addition to visualization of the viscera when the deep plicating sutures are taken, the pneumoperitoneum gives the surgeon a representative idea of what the reconstructed abdomen is going to look like when the patient is erect, thus enabling optimal abdominal wall reduction.
Outcome of Abdominoplasty Smith et all evaluated the voiding changes in twelve patients who underwent Monfort abdominoplasty. Subjective changes that occurred after abdominoplasty included resolution of or less double voiding in 9 patients, improved urinary continence in 7, improved bladder fullness sensation in 11, improved urinary flow in 10 and improved defecation in 5. They also noted that incidence of urinary tract infections decreased from a preoperative average of 5.7 per patient per year to 1.2 per patient per year postoperatively. In addition, post-void residual volumes significantly decreased from a preoperative average of 40% of bladder capacity to 13% after abdominoplasty even in those who did not undergo concomitant urinary reconstruction suggesting the influence of abdominoplasty on improved micturition.129 In the series by Denes et al, the flaccidity improved in 29 out of 30 patients who underwent comprehensive repair. Abdominoplasty improved not only the corporal image and self-esteem, but also abdominal strength, with good results in 93.5% of the patients. Upright body posture also improved in most patients. Although it is difficult to evaluate the cosmetic improvement objectively, this was substantiated by patient and parental satisfaction.112
Renal Transplantation Deteriorating renal function from renal dysplasia, recurrent pyelonephritis or reflux nephropathy are the reasons for renal replacement therapy. The incidence of renal transplantation in PBS patients is about 30%. 71 Graft survival after renal transplantation in PBS is similar to age matched controls, with graft survival of 75% versus 69% in controls. 138 Compared with age matched controls 10 years post transplant, improved survival of the renal graft was associated with pre-transplant urinary reconstruction to reduce the chance of urinary stasis and infection, as well as maintaining low post void residuals with timed voiding or clean intermittent catheterizations. 139 Most transplants have been carried out without139 prior abdominoplasty. When large urinary residue and UTI was encountered following transplantation, voiding efficiency and urinary infection improved following abdominoplasty.140 Torsion of the graft is a potential complication in PBS possibly due to lack of abdominal wall tone resulting in graft mobility.141 Prior abdominoplasty or nephropexy during transplantation have been recommended to avoid torsion.
Long-term Outlook
Because of the rarity of prune belly syndrome, most of the described literature is based on case series. Outcome of neonates of Category I, born with pulmonary dysplasia continues to be poor. In-hospital mortality of new-born patients with PBS dying during their initial hospital stay is 31%.Prematurity and pulmonary disorders were highly associated with in hospital mortality.10 With better neonatal care and efficient management of the urinary tract, survival has improved over the years. With individualized treatment plan and care, the overall outlook for the PBS patient, both for survival and for quality of care, has improved considerably, largely through advancements in neonatal care, surgical techniques and diagnostic evaluation. Though aggressive surgical approach may not be required in all patients, meticulous follow up and active management of pyelonephritis and urinary tract obstruction are essential. A third of those with impaired renal function at initial evaluation develop chronic renal insufficiency during childhood or adolescence. Outcome of renal transplantation seems comparable to age matched non-PBS patients. Bladder dynamics and voiding pattern also may change over time. Hence, long-term follow-up is essential to ensure near-normal quality of life for patients with prune belly syndrome.
Figure Legends
Figure 1: Lax abdominal wall seen in Prune Belly Syndrone
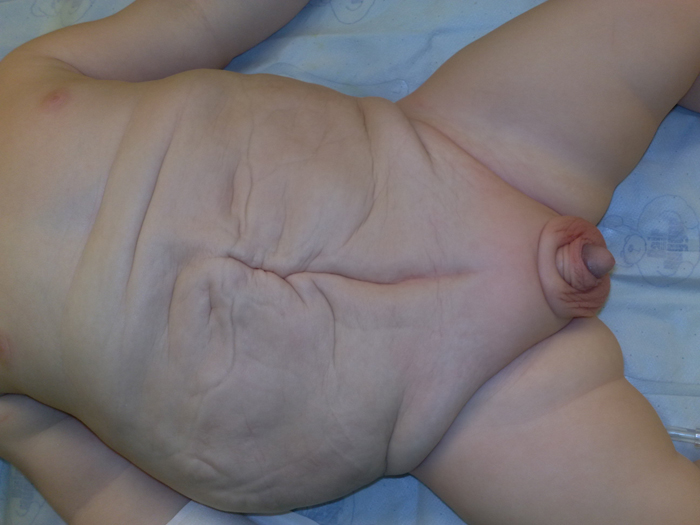
Figure 2: Renal Ultrasonogram of newborn with PBS with marked hydroureteronephrosis (Fig 2 A: Left kidney; Fig 2B: Right kidney)
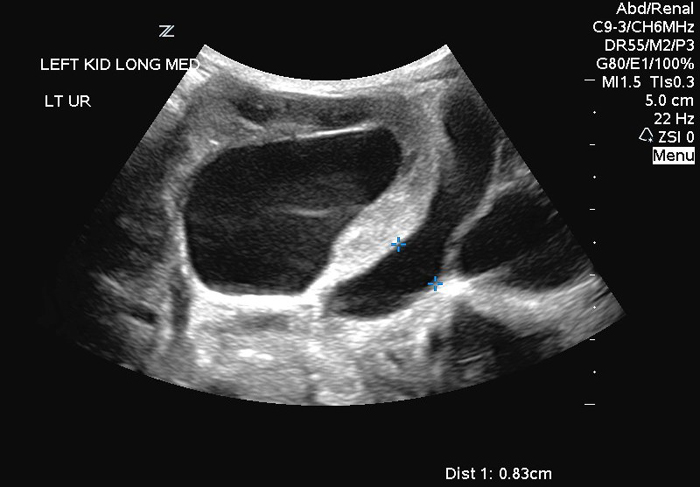
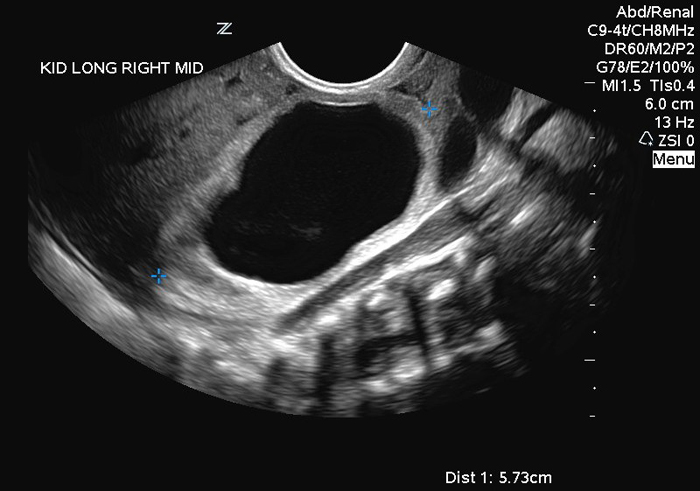
Figure 3: VCUG demonstrating bilateral vesicoureteral reflux with dilated ureters
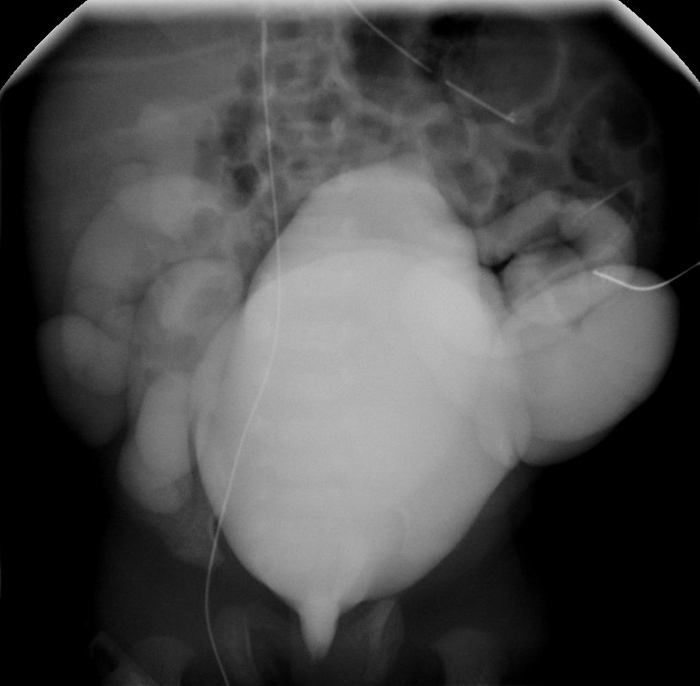
Figure 4: Ultrasonogram of the bladder demonstrating thickened bladder wall
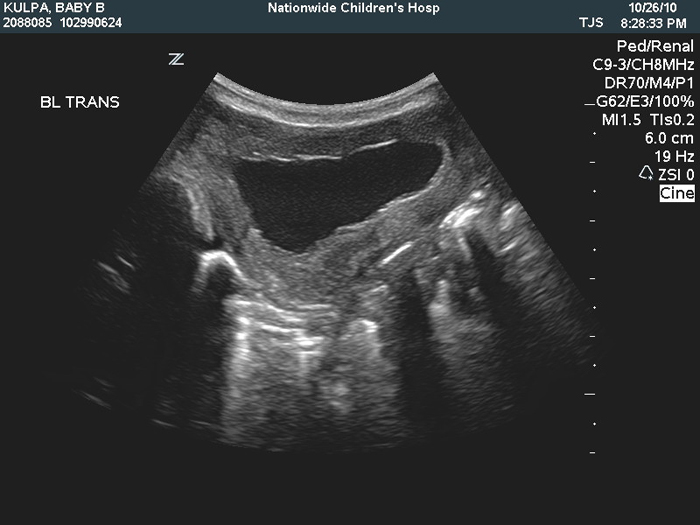
Figure 5: Hypoplastic prostate with dilated prostatic urethra
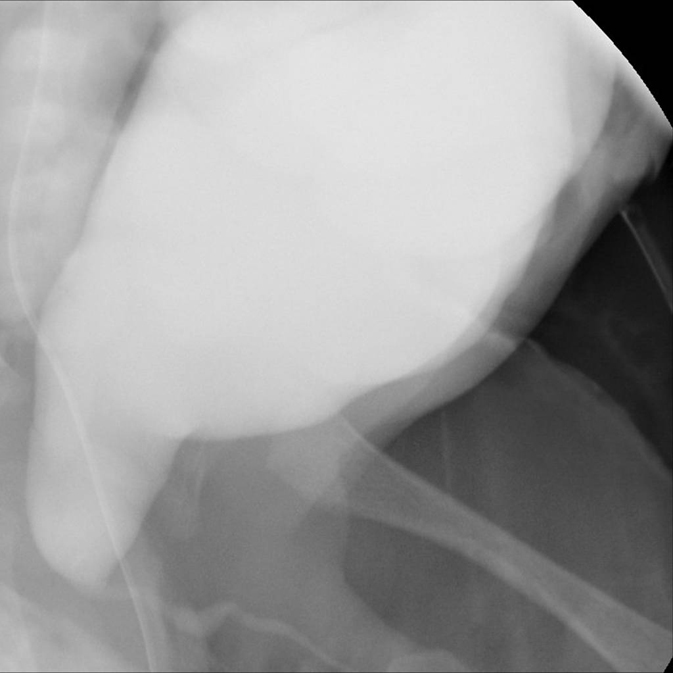
Figure 6: Cystogram demonstrating large bladder in a girl with PBS
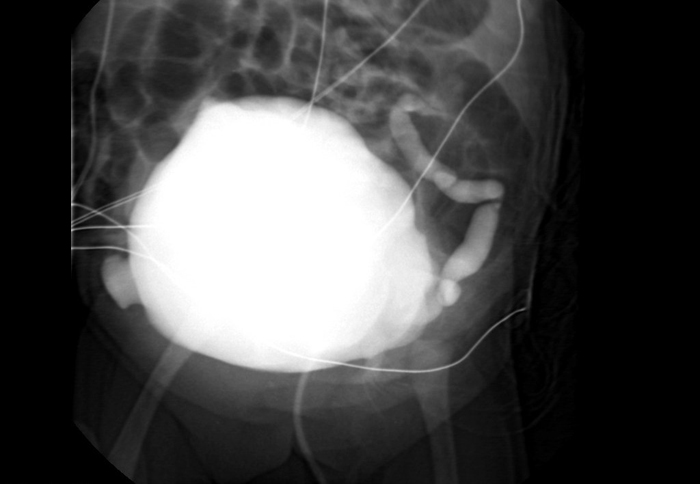
Figure 7: MAG 2 renogram in a boy with PBS, poorly functioning right kidney, dilated left collecting system and stasis of tracer.
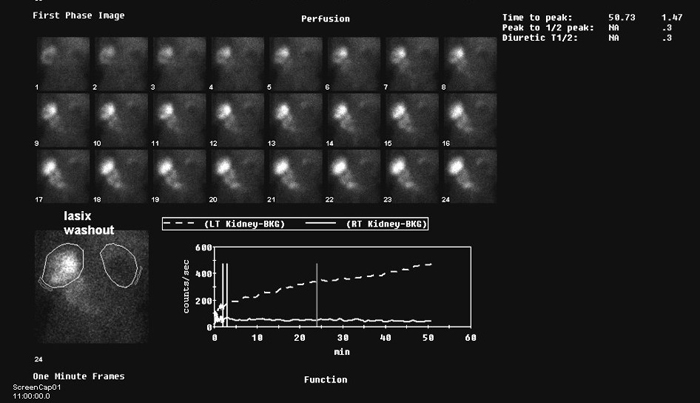
Figure 8 A: Incision marked for Montfort abdominoplasty. Skin island for neoumbilicus is also marked.
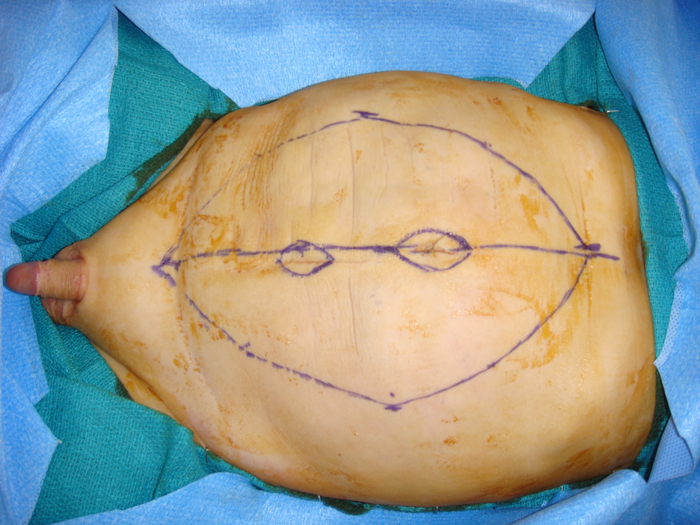
Figure 8 B: Skin flap has been raised preserving the vascularity to the skin islands for the neoumbilicus.
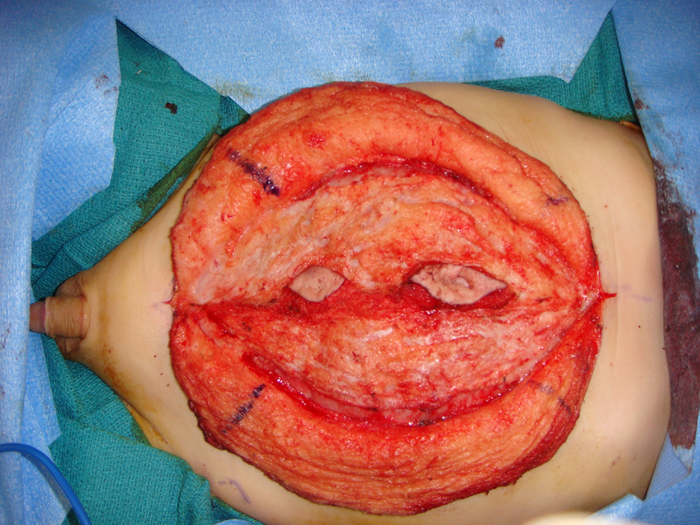
Figure 8 C: Scoring the peritoneum with diathermy,
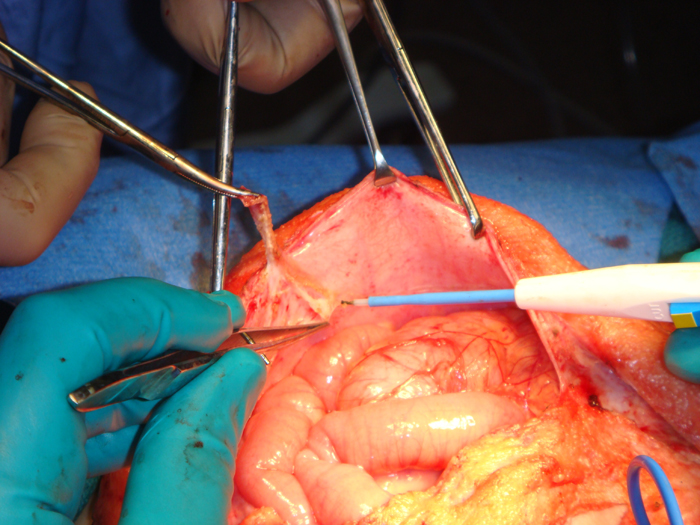
Figure 8 D: Securing the central island muscle flap to the posterior parietal peritoneum
References
1. Platt, W.: Rare Cause of Deficiency of the Abdominal Muscles. Philadelphia Medical Journal, 1: 738, 1898
2. Parker, R.: Absence of abdominal muscles in an infant. Lancet, 1: 1252, 1895
3. Osler, W.: Congenital absence of the abdominal muscles, with distended and hypertrophied urinary bladder. Bull Johns Hopkins Hospital, 12: 331, 1901
4. Nunn, I. N., Stephens, F. D.: The triad syndrome: a composite anomaly of the abdominal wall, urinary system and testes. J Urol, 86: 782, 1961
5. Ives, E. J.: The abdominal muscle deficiency triad syndrome--experience with ten cases. Birth Defects Orig Artic Ser, 10: 127, 1974
6. Eagle, J. F., Jr., Barrett, G. S.: Congenital deficiency of abdominal musculature with associated genitourinary abnormalities: A syndrome. Report of 9 cases. Pediatrics, 6: 721, 1950
7. Frydman, M., Magenis, R. E., Mohandas, T. K. et al.: Chromosome Abnormalities in infants with prune belly anomaly: association with trisomy 18. Am J Med Genet, 15: 145, 1983
8. Druschel, C. M.: A descriptive study of prune belly in New York State, 1983 to 1989. Arch Pediatr Adolesc Med, 149: 70, 1995
9. Wheatley, J. M., Stephens, F. D., Hutson, J. M.: Prune-belly syndrome: ongoing controversies regarding pathogenesis and management. Semin Pediatr Surg, 5: 95, 1996
10. Routh, J. C., Huang, L., Retik, A. B. et al.: Contemporary epidemiology and characterization of newborn males with prune belly syndrome. Urology, 76: 44, 2010
11. Reinberg, Y., Shapiro, E., Manivel, J. C. et al.: Prune belly syndrome in females: a triad of abdominal musculature deficiency and anomalies of the urinary and genital systems. J Pediatr, 118: 395, 1991
12. Ramasamy, R., Haviland, M., Woodard, J. R. et al.: Patterns of inheritance in familial prune belly syndrome. Urology, 65: 1227, 2005
13. Adeyokunnu, A. A., Familusi, J. B.: Prune belly syndrome in two siblings and a first cousin. Possible genetic implications. Am J Dis Child, 136: 23, 1982
14. Grenet, P., Le Calve, G., Badoual, J. et al.: [Congenital aplasia of the abdominal wall. A familial case]. Ann Pediatr (Paris), 19: 523, 1972
15. Harley, L. M., Chen, Y., Rattner, W. H.: Prune belly syndrome. J Urol, 108: 174, 1972
16. Afifi, A. K., Rebeiz, J., Mire, J. et al.: The myopathology of the Prune belly syndrome. J Neurol Sci, 15: 153, 1972
17. Garlinger, P., Ott, J.: Prune belly syndrome. Possible genetic implications. Birth Defects Orig Artic Ser, 10: 173, 1974
18. Riccardi, V. M., Grum, C. M.: The prune belly anomaly: heterogeneity and superficial X-linkage mimicry. J Med Genet, 14: 266, 1977
19. Lockhart, J. L., Reeve, H. R., Bredael, J. J. et al.: Siblings with prune belly syndrome and associated pulmonic stenosis, mental retardation, and deafness. Urology, 14: 140, 1979
20. Gaboardi, F., Sterpa, A., Thiebat, E. et al.: Prune-belly syndrome: report of three siblings. Helv Paediatr Acta, 37: 283, 1982
21. Feige, A., Fiedler, K., Rempen, A. et al.: [Prenatal diagnosis of prune belly syndrome occurring in siblings in 2 consecutive pregnancies]. Z Geburtshilfe Perinatol, 188: 239, 1984
22. Balaji, K. C., Patil, A., Townes, P. L. et al.: Concordant prune belly syndrome in monozygotic twins. Urology, 55: 949, 2000
23. Chan, Y. C., Bird, L. M.: Vertically transmitted hypoplasia of the abdominal wall musculature. Clin Dysmorphol, 13: 7, 2004
24. Frydman, M., Cohen, H. A., Ashkenazi, A. et al.: Familial segregation of cervical ribs, Sprengel anomaly, preaxial polydactyly, anal atresia, and urethral obstruction: a new syndrome? Am J Med Genet, 45: 717, 1993
25. Amacker, E. A., Grass, F. S., Hickey, D. E. et al.: An association of prune belly anomaly with trisomy 21. Am J Med Genet, 23: 919, 1986
26. Curry, C. J., Jensen, K., Holland, J. et al.: The Potter sequence: a clinical analysis of 80 cases. Am J Med Genet, 19: 679, 1984
27. Hoagland, M. H., Frank, K. A., Hutchins, G. M.: Prune-belly syndrome with prostatic hypoplasia, bladder wall rupture, and massive ascites in a fetus with trisomy 18. Arch Pathol Lab Med, 112: 1126, 1988
28. Salihu, H. M., Boos, R., Tchuinguem, G. et al.: Prenatal diagnosis of translocation and a single pericentric inversion 9: the value of fetal ultrasound. J Obstet Gynaecol, 21: 474, 2001
29. Fahmy, J., Kaminsky, C. K., Parisi, M. T.: Perlman syndrome: a case report emphasizing its similarity to and distinction from Beckwith-Wiedemann and prune-belly syndromes. Pediatr Radiol, 28: 179, 1998
30. Shah, D., Sharma, S., Faridi, M. M. et al.: VACTERL association with Prune-Belly syndrome. Indian Pediatr, 41: 845, 2004
31. Levin, T. L., Soghier, L., Blitman, N. M. et al.: Megacystis-microcolon-intestinal hypoperistalsis and prune belly: overlapping syndromes. Pediatr Radiol, 34: 995, 2004
32. Weber, S., Mir, S., Schlingmann, K. P. et al.: Gene locus ambiguity in posterior urethral valves/prune-belly syndrome. Pediatr Nephrol, 20: 1036, 2005
33. Sutherland, R. S., Mevorach, R. A., Kogan, B. A.: The prune-belly syndrome: current insights. Pediatr Nephrol, 9: 770, 1995
34. Pagon, R. A., Smith, D. W., Shepard, T. H.: Urethral obstruction malformation complex: a cause of abdominal muscle deficiency and the "prune belly". J Pediatr, 94: 900, 1979
35. Gonzalez, R., Reinberg, Y., Burke, B. et al.: Early bladder outlet obstruction in fetal lambs induces renal dysplasia and the prune-belly syndrome. J Pediatr Surg, 25: 342, 1990
36. Lattimer, J. K.: Congenital deficiency of the abdominal musculature and associated genitourinary anomalies: a report of 22 cases. J Urol, 79: 343, 1958
37. Rogers, L. W., Ostrow, P. T.: The prune belly syndrome. Report of 20 cases and description of a lethal variant. J Pediatr, 83: 786, 1973
38. Moerman, P., Fryns, J. P., Goddeeris, P. et al.: Pathogenesis of the prune-belly syndrome: a functional urethral obstruction caused by prostatic hypoplasia. Pediatrics, 73: 470, 1984
39. Rich, D.: The Prune Belly Syndrome. AUA Update Series, 11: 721, 1992
40. Mininberg, D. T., Montoya, F., Okada, K. et al.: Subcellular muscle studies in the prune belly syndrome. J Urol, 109: 524, 1973
41. O'Kell, R. T.: Embryonic abdominal musculature associated with anomalies of the genitourinary and gastrointestinal systems. Am J Obstet Gynecol, 105: 1283, 1969
42. Stephens, F. D., Gupta, D.: Pathogenesis of the prune belly syndrome. J Urol, 152: 2328, 1994
43. Wigger, H. J., Blanc, W. A.: The prune belly syndrome. Pathol Annu, 12 Pt 1: 17, 1977
44. Deklerk, D. P., Scott, W. W.: Prostatic maldevelopment in the prune belly syndrome: a defect in prostatic stromal-epithelial interaction. J Urol, 120: 341, 1978
45. Elder, J. S., Isaacs, J. T., Walsh, P. C.: Androgenic sensitivity of the gubernaculum testis: evidence for hormonal/mechanical interactions in testicular descent. J Urol, 127: 170, 1982
46. Fallon, B., Welton, M., Hawtrey, C.: Congenital anomalies associated with cryptorchidism. J Urol, 127: 91, 1982
47. Reinberg, Y., Manivel, J. C., Pettinato, G. et al.: Development of renal failure in children with the prune belly syndrome. J Urol, 145: 1017, 1991
48. Burke, E. C., Shin, M. H., Kelalis, P. P.: Prune-belly syndrome. Clinical findings and survival. Am J Dis Child, 117: 668, 1969
49. Christopher, C. R., Spinelli, A., Severt, D.: Ultrasonic diagnosis of prune-belly syndrome. Obstet Gynecol, 59: 391, 1982
50. Hudson, R., Skoog, S.: Prune Belly Syndrome, 5 ed, 2007
51. Jennings, R. W.: Prune belly syndrome. Semin Pediatr Surg, 9: 115, 2000
52. Woodard, J. R.: The prune belly syndrome. Urol Clin North Am, 5: 75, 1978
53. Caldamone, A., Woodard, J. R.: Prune Belly Syndrome, 2009
54. Manivel, J. C., Pettinato, G., Reinberg, Y. et al.: Prune belly syndrome: clinicopathologic study of 29 cases. Pediatr Pathol, 9: 691, 1989
55. Gearhart, J. P., Lee, B. R., Partin, A. W. et al.: A quantitative histological evaluation of the dilated ureter of childhood. II: Ectopia, posterior urethral valves and the prune belly syndrome. J Urol, 153: 172, 1995
56. Palmer, J. M., Tesluk, H.: Ureteral pathology in the prune belly syndrome. J Urol, 111: 701, 1974
57. Workman, S. J., Kogan, B. A.: Fetal bladder histology in posterior urethral valves and the prune belly syndrome. J Urol, 144: 337, 1990
58. Snyder, H. M., Harrison, N. W., Whitfield, H. N. et al.: Urodynamics in the prune belly syndrome. Br J Urol, 48: 663, 1976
59. Kinahan, T. J., Churchill, B. M., McLorie, G. A. et al.: The efficiency of bladder emptying in the prune belly syndrome. J Urol, 148: 600, 1992
60. Williams, D. I., Burkholder, G. V.: The prune belly syndrome. J Urol, 98: 244, 1967
61. Passerini-Glazel, G., Araguna, F., Chiozza, L. et al.: The P.A.D.U.A. (progressive augmentation by dilating the urethra anterior) procedure for the treatment of severe urethral hypoplasia. J Urol, 140: 1247, 1988
62. Shrom, S. H., Cromie, W. J., Duckett, J. W., Jr.: Megalourethra. Urology, 17: 152, 1981
63. Kroovand, R. L., Al-Ansari, R. M., Perlmutter, A. D.: Urethral and genital malformations in prune belly syndrome. J Urol, 127: 94, 1982
64. Cabral, B., Majidi, A., Gonzalez, R.: Ectopic vasa deferentia in an infant with the prune belly syndrome. J Urol (Paris), 94: 223, 1988
65. Orvis, B. R., Bottles, K., Kogan, B. A.: Testicular histology in fetuses with the prune belly syndrome and posterior urethral valves. J Urol, 139: 335, 1988
66. Massad, C. A., Cohen, M. B., Kogan, B. A. et al.: Morphology and histochemistry of infant testes in the prune belly syndrome. J Urol, 146: 1598, 1991
67. Batata, M. A., Chu, F. C., Hilaris, B. S. et al.: Testicular cancer in cryptorchids. Cancer, 49: 1023, 1982
68. Sayre, R., Stephens, R., Chonko, A. M.: Prune belly syndrome and retroperitoneal germ cell tumor. Am J Med, 81: 895, 1986
69. Woodhouse, C. R., Ransley, P. G.: Teratoma of the testis in the prune belly syndrome. Br J Urol, 55: 580, 1983
70. Woodhouse, C. R., Snyder, H. M., 3rd: Testicular and sexual function in adults with prune belly syndrome. J Urol, 133: 607, 1985
71. Burbige, K. A., Amodio, J., Berdon, W. E. et al.: Prune belly syndrome: 35 years of experience. J Urol, 137: 86, 1987
72. Kolettis, P. N., Ross, J. H., Kay, R. et al.: Sperm retrieval and intracytoplasmic sperm injection in patients with prune-belly syndrome. Fertil Steril, 72: 948, 1999
73. Holder, J. P.: Pathophysiologic and anesthetic correlations of the prune-belly syndrome. AANA J, 57: 137, 1989
74. Straub, E., Spranger, J.: Etiology and pathogenesis of the prune belly syndrome. Kidney Int, 20: 695, 1981
75. Brinker, M. R., Palutsis, R. S., Sarwark, J. F.: The orthopaedic manifestations of prune-belly (Eagle-Barrett) syndrome. J Bone Joint Surg Am, 77: 251, 1995
76. Carey, J. C., Eggert, L., Curry, C. J.: Lower limb deficiency and the urethral obstruction sequence. Birth Defects Orig Artic Ser, 18: 19, 1982
77. Joller, R., Scheier, H.: Complete thoracic segmental insensibility accompanying prune belly syndrome with scoliosis. Spine (Phila Pa 1976), 11: 496, 1986
78. Geary, D. F., MacLusky, I. B., Churchill, B. M. et al.: A broader spectrum of abnormalities in the prune belly syndrome. J Urol, 135: 324, 1986
79. Wright, J. R., Jr., Barth, R. F., Neff, J. C. et al.: Gastrointestinal malformations associated with prune belly syndrome: three cases and a review of the literature. Pediatr Pathol, 5: 421, 1986
80. Teramoto, R., Opas, L. M., Andrassy, R.: Splenic torsion with prune belly syndrome. J Pediatr, 98: 91, 1981
81. Short, K. L., Groff, D. B., Cook, L.: The concomitant presence of gastroschisis and prune belly syndrome in a twin. J Pediatr Surg, 20: 186, 1985
82. Walker, J., Prokurat, A. I., Irving, I. M.: Prune belly syndrome associated with exomphalos and anorectal agenesis. J Pediatr Surg, 22: 215, 1987
83. Salihu, H. M., Tchuinguem, G., Aliyu, M. H. et al.: Prune belly syndrome and associated malformations. A 13-year experience from a developing country. West Indian Med J, 52: 281, 2003
84. Woodard, J. R., Parrott, T. S.: Reconstruction of the urinary tract in prune belly uropathy. J Urol, 119: 824, 1978
85. Ewig, J. M., Griscom, N. T., Wohl, M. E.: The effect of the absence of abdominal muscles on pulmonary function and exercise. Am J Respir Crit Care Med, 153: 1314, 1996
86. Alford, B. A., Peoples, W. M., Resnick, J. S. et al.: Pulmonary complications associated with the prune-belly syndrome. Radiology, 129: 401, 1978
87. Bellah, R. D., States, L. J., Duckett, J. W.: Pseudoprune-Belly syndrome: imaging findings and clinical outcome. AJR Am J Roentgenol, 167: 1389, 1996
88. Yamamoto, H., Nishikawa, S., Hayashi, T. et al.: Antenatal diagnosis of prune belly syndrome at 11 weeks of gestation. J Obstet Gynaecol Res, 27: 37, 2001
89. Ellison, L., Cendron, M., Ornvold, K. et al.: Early diagnosis of fetal bladder outlet obstruction. J Pediatr Surg, 35: 513, 2000
90. Elder, J.: Intrauterine intervention for obstructive uropathy. Kidney 22: 19, 1990
91. Gadziala, N. A., Kawada, C. Y., Doherty, F. J. et al.: Intrauterine decompression of megalocystis during the second trimester of pregnancy. Am J Obstet Gynecol, 144: 355, 1982
92. Glazer, G. M., Filly, R. A., Callen, P. W.: The varied sonographic appearance of the urinary tract in the fetus and newborn with urethral obstruction. Radiology, 144: 563, 1982
93. Nakayama, D. K., Harrison, M. R., Gross, B. H. et al.: Management of the fetus with an abdominal wall defect. J Pediatr Surg, 19: 408, 1984
94. Leeners, B., Sauer, I., Schefels, J. et al.: Prune-belly syndrome: therapeutic options including in utero placement of a vesicoamniotic shunt. J Clin Ultrasound, 28: 500, 2000
95. Pescia, G., Cruz, J. M., Weihs, D.: Prenatal diagnosis of prune belly syndrome by means of raised maternal AFP levels. J Genet Hum, 30: 271, 1982
96. McLorie, G., Farhat, W., Khoury, A. et al.: Outcome analysis of vesicoamniotic shunting in a comprehensive population. J Urol, 166: 1036, 2001
97. Freedman, A. L., Johnson, M. P., Smith, C. A. et al.: Long-term outcome in children after antenatal intervention for obstructive uropathies. Lancet, 354: 374, 1999
98. Kramer, S. A.: Current status of fetal intervention for congenital hydronephrosis. J Urol, 130: 641, 1983
99. Makino, Y., Kobayashi, H., Kyono, K. et al.: Clinical results of fetal obstructive uropathy treated by vesicoamniotic shunting. Urology, 55: 118, 2000
100. Irwin, B. H., Vane, D. W.: Complications of intrauterine intervention for treatment of fetal obstructive uropathy. Urology, 55: 774, 2000
101. Soylu, H., Kutlu, N. O., Sonmezgoz, E. et al.: Prune-belly syndrome and pulmonary hypoplasia: a potential cause of death. Pediatr Int, 43: 172, 2001
102. Noh, P. H., Cooper, C. S., Winkler, A. C. et al.: Prognostic factors for long-term renal function in boys with the prune-belly syndrome. J Urol, 162: 1399, 1999
103. Perlman, M., Levin, M.: Fetal pulmonary hypoplasia, anuria, and oligohydramnios: clinicopathologic observations and review of the literature. Am J Obstet Gynecol, 118: 1119, 1974
104. Woodard, J. R.: Lessons learned in 3 decades of managing the prune-belly syndrome. J Urol, 159: 1680, 1998
105. Berdon, W. E., Baker, D. H., Wigger, H. J. et al.: The radiologic and pathologic spectrum of the prune belly syndrome. The importance of urethral obstruction in prognosis. Radiol Clin North Am, 15: 83, 1977
106. Fallat, M. E., Skoog, S. J., Belman, A. B. et al.: The prune belly syndrome: a comprehensive approach to management. J Urol, 142: 802, 1989
107. Woodard, J. R.: Prune belly syndrome. In: Clinical Pediatric Urology. Edited by K. L. R. Kelalis P.P., Belman A.B. Philadelphia: WB Saunders, pp. 805-824, 1985
108. Waldbaum, R. S., Marshall, V. F.: The prune belly syndrome: a diagnostic therapeutic plan. J Urol, 103: 668, 1970
109. Woodhouse, C. R., Kellett, M. J., Williams, D. I.: Minimal surgical interference in the prune belly syndrome. Br J Urol, 51: 475, 1979
110. Randolph, J., Cavett, C., Eng, G.: Surgical correction and rehabilitation for children with "Prune-belly" syndrome. Ann Surg, 193: 757, 1981
111. Woodhouse, C. R., Ransley, P. G., Innes-Williams, D.: Prune belly syndrome--report of 47 cases. Arch Dis Child, 57: 856, 1982
112. Denes, F. T., Arap, M. A., Giron, A. M. et al.: Comprehensive surgical treatment of prune belly syndrome: 17 years' experience with 32 patients. Urology, 64: 789, 2004
113. Tank, E. S., McCoy, G.: Limited surgical intervention in the prune belly syndrome. J Pediatr Surg, 18: 688, 1983
114. Randolph, J. G.: Total surgical reconstruction for patients with abdominal muscular deficiency ("prune-belly") syndrome. J Pediatr Surg, 12: 1033, 1977
115. Woodard, J. R.: Prune-belly syndrome: a personal learning experience. BJU Int, 92 Suppl 1: 10, 2003
116. Perlmutter, A. D.: Reduction cystoplasty in prune belly syndrome. J Urol, 116: 356, 1976
117. Bukowski, T. P., Perlmutter, A. D.: Reduction cystoplasty in the prune belly syndrome: a long-term followup. J Urol, 152: 2113, 1994
118. Gonzalez, R., De Filippo, R., Jednak, R. et al.: Urethral atresia: long-term outcome in 6 children who survived the neonatal period. J Urol, 165: 2241, 2001
119. Reinberg, Y., Chelimsky, G., Gonzalez, R.: Urethral atresia and the prune belly syndrome. Report of 6 cases. Br J Urol, 72: 112, 1993
120. Jones, E. A., Freedman, A. L., Ehrlich, R. M.: Megalourethra and urethral diverticula. Urol Clin North Am, 29: 341, 2002
121. Heaton, B. W., Snow, B. W., Cartwright, P. C.: Repair of urethral diverticulum by plication. Urology, 44: 749, 1994
122. Locke, J. R., Noe, H. N.: Megalourethra: surgical technique for correction of an unusual variant. J Urol, 138: 110, 1987
123. Stephens, F. D.: Congenital malformations of the urinary tract. New York: Praeger, pp. xiv, 577 p., 1983
124. Smith, E. A., Woodard, J. R.: Prune Belly Syndrome. In: Campbell's Urology, 8 ed. Edited by P. C. Walsh, A. J. Wein, E. D. Vaughan et al. Philadelphia: Saunders, vol. 2, 2002
125. Woodard, J. R., Parrott, T. S.: Orchiopexy in the prune belly syndrome. Br J Urol, 50: 348, 1978
126. Docimo, S. G., Moore, R. G., Kavoussi, L. R.: Laparoscopic orchidopexy in the prune belly syndrome: a case report and review of the literature. Urology, 45: 679, 1995
127. Patil, K. K., Duffy, P. G., Woodhouse, C. R. et al.: Long-term outcome of Fowler-Stephens orchiopexy in boys with prune-belly syndrome. J Urol, 171: 1666, 2004
128. Boddy, S. A., Gordon, A. C., Thomas, D. F. et al.: Experience with the Fowler Stephens and microvascular procedures in the management of intraabdominal testes. Br J Urol, 68: 199, 1991
129. Smith, C. A., Smith, E. A., Parrott, T. S. et al.: Voiding function in patients with the prune-belly syndrome after Monfort abdominoplasty. J Urol, 159: 1675, 1998
130. Randolph, J., Cavett, C., Eng, G.: Abdominal wall reconstruction in the prune belly syndrome. J Pediatr Surg, 16: 960, 1981
131. Ehrlich, R. M., Lesavoy, M. A., Fine, R. N.: Total abdominal wall reconstruction in the prune belly syndrome. J Urol, 136: 282, 1986
132. Ehrlich, R. M., Lesavoy, M. A.: Umbilicus preservation with total abdominal wall reconstruction in prune-belly syndrome. Urology, 41: 231, 1993
133. Monfort, G., Guys, J. M., Bocciardi, A. et al.: A novel technique for reconstruction of the abdominal wall in the prune belly syndrome. J Urol, 146: 639, 1991
134. Parrott, T. S., Woodard, J. R.: The Monfort operation for abdominal wall reconstruction in the prune belly syndrome. J Urol, 148: 688, 1992
135. Bukowski, T. P., Smith, C. A.: Monfort abdominoplasty with neoumbilical modification. J Urol, 164: 1711, 2000
136. Furness, P. D., 3rd, Cheng, E. Y., Franco, I. et al.: The prune-belly syndrome: a new and simplified technique of abdominal wall reconstruction. J Urol, 160: 1195, 1998
137. Levine, E., Taub, P. J., Franco, I.: Laparoscopic-assisted abdominal wall reconstruction in prune-belly syndrome. Ann Plast Surg, 58: 162, 2007
138. Reinberg, Y., Manivel, J. C., Fryd, D. et al.: The outcome of renal transplantation in children with the prune belly syndrome. J Urol, 142: 1541, 1989
139. Kamel, M. H., Thomas, A. A., Al-Mufarrej, F. M. et al.: Deceased-donor kidney transplantation in prune belly syndrome. Urology, 69: 666, 2007
140. Fusaro, F., Zanon, G. F., Ferreli, A. M. et al.: Renal transplantation in prune-belly syndrome. Transpl Int, 17: 549, 2004
141. Marvin, R. G., Halff, G. A., Elshihabi, I.: Renal allograft torsion associated with prune-belly syndrome. Pediatr Nephrol, 9: 81, 1995








