









Síndrome de prune belly
J CHANDRA SINGH, MD1, DOUGLAS W STORM, MD2, ASHAY PATEL, DO1, VENKATA RAMA JAYANTHI, MD1
1División de Urología Pediátrica, Hospital Nacional para Niños, Columbus, OH, USA
[email protected]
2Departamento de Urología, San Diego Naval Medical Center, San Diego, CA, USA
[email protected]
Traducido y editado desde el original al español | Enlace a la versión en inglés
Francisca Yankovic
Hospital Exequiel Gonzalez Cortes, Santiago Chile
INTRODUCCIÓN
El Síndrome de Prune Belly (SPB) consiste en una constelación de malformaciones congénitas, con las tres anomalías principales que son: laxitud de la pared abdominal, testículos bilaterales no descendidos y anomalías del tracto genitourinario. Si bien la apariencia inusual de abdomen del tipo "ciruela" (Figura 1) es el sello que generalmente identifica a estos pacientes, la función renal es el factor más importante para determinar su supervivencia general. Las anomalías urinarias que se han asociado con el síndrome del vientre de ciruela incluyen: hidronefrosis, uréteres dilatados tortuosos, grados variables de displasia renal y mega vejiga. Los sistemas respiratorio, musculo esquelético, gastrointestinal, ortopédico y cardíaco también pueden estar afectados.
El defecto de la pared abdominal fue descrito por primera vez por Frölich en 1839 y más tarde fue registrado gráficamente por Platt.1 Parker fue el primero en informar sobre un bebé varón con los tres componentes clásicos del síndrome de SPB. 2 En 1901, Osler fue el primero en utilizar el nombre "vientre de ciruela pasa" y reconoció la vejiga distendida asociada con este síndrome.1,3 Si bien, el título de "síndrome de vientre de ciruela pasa" ha persistido a lo largo de los años, otros han encontrado esta designación de mal gusto y han utilizado nombres como el "síndrome de la tríada" "Displasia mesenquimatosa" y el "síndrome de Eagle-Barrett". 4-6
El síndrome de SPB ocurre entre 1 en 30,000 a 1 en 50,000 nacimientos, y afecta en un 95% a bebés varones.7-9 Una reciente revisión de la base de datos de pacientes hospitalizados reveló 133 bebés varones recién nacidos con SBP de 1 420 991 nacimientos, con una estimación de la incidencia de 3.8 casos / 100 000 nacidos vivos10. Las mujeres afectadas presentan ausencia de musculatura abdominal y anomalías de los sistemas genital y urinario11. También parece haber una mayor incidencia en embarazos gemelares, así como en niños nacidos de madres más jóvenes8.
GENÉTICA
No existe un patrón genético claro para el SPB, aunque hay un patrón de herencia familiar hipotético en algunos casos aislados. Se ha informado ocurrencia de SPB en gemelos, primos y hermanos, lo que sugiere una herencia familiar.12-23 Ramasamy y otros revisaron la mayoría de estos casos familiares en 2004.12 Supusieron que podría haber un patrón de herencia autosómica recesiva que podría explicar los casos aislados. Como las enfermedades heredadas en un patrón autosómico recesivo parecen ocurrir con mayor frecuencia en los matrimonios consanguíneos, los informes de Frydman y Adeyokunnu13,24 reportan un caso de SPB en un varón nacido una pareja en una relación consanguínea; mientras que Adeyokunnu informó una mayor incidencia de este síndrome en Nigeria, un área con mayores tasas conocidas de consanguinidad13,24. Riccardi y Grum también han sugerido una mutación autosómica dominante de dos pasos con expresión limitada ligada al sexo que puede explicar el predominio masculino de este síndrome, así como el número poco frecuente de miembros una familia afectados.18 Si bien estos reportes constituyen un argumento para un patrón de herencia familiar, la mayoría de los casos parecen ocurrir de manera esporádica. En su revisión de Ives et al., los niños afectados con el SPB describieron once pares de gemelos discordantes para la expresión del síndrome, lo que habla en contra de una base completamente genética. 24
Además, las anomalías cromosómicas específicas también son raras. El SPB se asoció con trisomía 13, trisomía 18, trisomía 21 y síndrome de Turner, pero no se ha demostrado una relación específica. 25-27 También se han notificado translocaciones e inversiones peri céntricas del cromosoma 9 en los fetos que presentan este síndrome. Sin embargo, estos hallazgos cromosómicos son inespecíficos, ya que no todos los niños con este síndrome tienen estas anomalías cromosómicas, ni todos los niños con inversiones peri céntricas del cromosoma 9 presentan el SPB 28.
Se han descrito asociaciones entre las válvulas uretrales posteriores, el síndrome de Perlman, el síndrome de Beckwith-Wiedemann, la asociación VACTERL y el síndrome de hipoperistalsis megacystis-microcolon-intestinal.29-32 Estas asociaciones sugieren una patogenia común entre el SPB y estas otras condiciones.
EMBRIOLOGÍA
Existen tres teorías principales que explican el SPB: obstrucción fetal al tracto de salida, injuria de mesodermo y la teoría del saco vitelino.
Obstrucción fetal al tracto salida
En 1903, Stumme sugirió que el SPB pasa por una obstrucción de la salida de la vejiga.33 Supuso que esta obstrucción infra vesical provocaba la distensión de la vejiga, la dilatación uretral y la hidronefrosis características de este síndrome. También se consideró que la dilatación de la vejiga daba como resultado atrofia de la pared abdominal a través del infarto venoso y también obstruía el descenso normal de los testículos.34 Esta teoría fue luego corroborada por González y otros, quienes recrearon los hallazgos fenotípicos encontrados en el SPB mediante obstrucción la uretra fetal de cordero a los 43-45 días de gestación.35 Si la obstrucción infra vesical resultará o no en los hallazgos asociados con el SPB pasa por el momento en que ocurre y la gravedad de esta obstrucción, ya que los hallazgos asociados de este síndrome son distintos de otras uropatías obstructivas. Según la línea de tiempo de desarrollo del feto, parece que la obstrucción debería ocurrir entre las semanas 13 y 15 de gestación. En este momento, la obstrucción podría explicar las anomalías encontradas en el SPB ya que, en ese momento, el uraco ha comenzado a cerrarse, se ha producido una importante producción de orina fetal y el desarrollo prostático también está comenzando9.
En los seres humanos, la obstrucción uretral en realidad solo se identifica en aproximadamente el 10-20% de los casos.36 Algunos han postulado que la obstrucción puede ser transitoria, mientras que otros han especulado que las personas con lesiones obstructivas solo pueden representar la forma más grave del SPB. La mayoría de estos bebés que realmente demuestran obstrucción uretral provienen de estudios de autopsia, por lo tanto, algunos creen que el modelo de obstrucción uretral puede explicar "la variante letal" y puede que no cuente con niños que sobreviven.37 Para contrarrestar este argumento, algunos han supuesto que la uretra prostática hipoplásica podría funcionar como una flap valvular obstructivo. En una serie de autopsias, Moerman et al descubrieron que las próstatas de los niños con SPB carecían de fibras musculares lisas y que las glándulas tuboalveolares estaban reducidas en número. 38 Ellos postularon que estas próstatas hipoplásicas resultaron en debilidad de las paredes prostáticas y en la saculación de la uretra prostática. Notaron que este abultamiento era más marcado a lo largo de los aspectos dorsal y caudal, y consideraron que esto hacía que la uretra membranosa se insertara más dorsalmente, de manera oblicua, en la uretra prostática. Se consideró que esta inserción anormal en la uretra prostática hipoplásica podría dar lugar a un mecanismo valvular, causando una obstrucción funcional. De esta manera, la hipoplasia prostática podría ser la causa y no necesariamente el efecto del SPB. Otros han planteado la hipótesis de que la canalización tardía de la uretra distal podría ser la causa de obstrucción.39 Este retraso podría explicar la incapacidad de identificar una verdadera obstrucción en el momento del nacimiento.
La teoría obstructiva sostiene que los hallazgos de la pared abdominal son secundarios a los efectos de la presión de la vejiga en los músculos abdominales en desarrollo. Apoyando esta teoría, los músculos directamente sobre la vejiga parecen ser los más afectados. Además, Moerman et al también identificaron cambios musculares distróficos tras el análisis histológico en su estudio de autopsia.38 Consideraron que estos hallazgos histológicos podrían ser compatibles con la descripción de Pinto quien describe el infarto venoso muscular secundario a la presión de la vejiga distendida. Sin embargo, otros estudios histológicos y de microscopía electrónica de la pared abdominal son más consistentes con la detención del desarrollo, más que la atrofia, y no apoyan la obstrucción de la salida como causa de anomalías de la pared abdominal40,41.
Si bien la obstrucción infra vesical fetal parece plausible, no tiene en cuenta todos los hallazgos asociados con el SPB. Ciertamente, hay inconsistencias con esta teoría y no todos los factores comunes en los niños con este síndrome pueden ser explicados por este modelo.
Teoría de la injuria Mesodérmica
Esta teoría sugiere que la etiología del SPB pasa por un insulto nocivo al mesodermo embrionario. Durante el desarrollo fetal normal, el mesodermo fluye hacia afuera entre el ectodermo y el endodermo. El mesodermo luego se divide en segmentos medial y lateral, que forman los somitos y la placa lateral, respectivamente. La placa lateral luego se divide en una capa visceral y somática. La capa somática entre los somitas y la placa lateral forma el mesodermo nefrogénico, que es el precursor del mesonefros, el conducto de Wolff y el metanefros. La capa somática del mesodermo de la placa lateral es la fuente de la musculatura de la pared abdominal, mientras que la capa visceral del mesodermo de la placa lateral da lugar al músculo liso de los tractos urinario y gastrointestinal.
Stephens y Gupta propusieron que las anomalías del tracto urinario podrían explicarse por el desarrollo anormal de los mesonefros entre las semanas 6 y 10. Normalmente, el conducto de Wolff, que es un derivado del mesodermo, termina en su porción caudal en el seno urogenital y se incorpora a la uretra membranosa futura y delimita el verumontanum. Los conductos de Wolff y las yemas ureterales se incorporan adicionalmente en el trígono, con las yemas ureterales crecen hacia el blastema nefrogénico. La incorporación anormal de los conductos wolffianos terminales podría producir hipoplasia prostática, dilatación de la uretra prostática e incluso obstrucciones similares a válvulas de uretra posterior. Adicionalmente, Ives propuso que el defecto mesodérmico podría ocurrir tan precoz como en la tercera semana de gestación, durante la división del disco embrionario, que normalmente ocurre en esta semana, lo que podría explicar la aparición de gemelos monocigóticos discordantes.5 Otros postulan que las anomalías mesodérmicas también podrían explicar las alteraciones del descenso testicular, ya que una anomalía en los gubernáculos testiculares, también derivados del mesodermo, provocarían una falta de respuesta de estos tejidos a las influencias hormonales y, por lo tanto, impedirían que los testículos desciendan normalmente.43-46 Sin embargo, la teoría de la injuria mesodérmica no explica todos los factores asociados con el SPB. Se necesitan más pruebas y explicaciones para apoyar completamente esta teoría como la causa de este síndrome.
La teoría del saco vitelino
Stephens ha propuesto una teoría adicional para explicar las características asociadas con el SPB. Su hipótesis es que, en los niños con este síndrome, la vejiga y la uretra posterior derivan de una porción más grande de la alantoides y que retienen una cantidad anormal del saco vitelino. Esta cantidad anormal de saco vitelino podría afectar el desarrollo de la pared abdominal y dar como resultado una apariencia de "ciruela". Además, el divertículo alantoico puede incorporarse en el tracto urinario y explicar el uraco, vejiga y la uretra prostática agrandados propios de este síndrome. 42 Sin embargo, esta teoría no tiene en cuenta todas las anomalías observadas en el riñón y tracto genital.
CARACTERÍSTICAS CLÍNICAS
Anormalidades genitourinarias
Riñones
Existe una amplia variación en la presentación renal, que va desde los riñones normales hasta los no funcionales displásicos severos.47 Es el grado de displasia renal y la función renal general del niño lo que generalmente determina su pronóstico. Los problemas renales y la hipoplasia pulmonar resultante dan cuenta del 20% de posibilidades de muerte fetal en el período neonatal, y un 30% adicional de progresión a la insuficiencia renal terminal en los primeros dos años de vida.33,48 Histológicamente, los riñones más displásicos son similares a los descritos por Potter que demuestran pocas nefronas y otras características de desorganización del parénquima.
La hidronefrosis también es un hallazgo común (Figura 2A, B) y en la actualidad generalmente se detecta en las ecografías prenatales.49 Hay que recordar que el grado de hidronefrosis no se correlaciona necesariamente con la función renal, el grado de dilatación ureteral o la laxitud de la pared abdominal50. Además, no es inusual identificar compromiso renal asimétrico, ya que un riñón muestra hidronefrosis y / o displasia severas en comparación con el lado contralateral normal.51 También se ha informado obstrucción de la unión ureteropélvica, sin embargo, la hidronefrosis no obstructiva es lo más frecuente. 52 En aquellos pacientes con función renal preservada, parece que las infecciones del tracto urinario pueden representar el mayor riesgo de dañar sus riñones.53
Uréteres
Los uréteres son alargados, dilatados y tortuosos (Figura 3); el tercio inferior está más profundamente afectado en comparación con la porción proximal.33 El reflujo vesicoureteral está presente hasta en el 75% de los pacientes y los orificios uretéricos pueden ser amplios y patulosos. 9 Las imágenes fluoroscópicas de los uréteres pueden mostrar peristalsis inefectiva, sin embargo, típicamente no están obstruidas.33 Aunque son poco frecuentes, existen informes de obstrucción tanto en la unión ureteropélvica como en la unión ureterovesical.38,43,54
El examen histológico de los uréteres revela alteraciones en la arquitectura de la pared ureteral. La hipertrofia y la hiperplasia del músculo liso, que se pueden observar en pacientes con válvulas uretrales posteriores y reflujo vesicoureteral, generalmente no se presentan en el SPB. En cambio, el músculo liso es escaso o incluso ausente y puede ser reemplazado por tejido fibroso celular.4,38 También parece haber un aumento en la proporción de colágeno a músculo liso en la capa muscular uretérica, especialmente si hay reflujo vesicoureteral .55 Aunque está presente en todo el uréter, la porción proximal del uréter parece tener más células musculares lisas en comparación con la porción distal.4,56 También parece haber una disminución en el número de plexos nerviosos y fibras de Schwann dentro del uréter. Estas características histológicas pueden interferir con la propagación de impulsos de célula a célula y dar lugar a una peristalsis ureteral deficiente. Estas características también deben tenerse en cuenta si se realiza cirugía ureteral en estos pacientes.
Vejiga
La vejiga en pacientes con el SPB generalmente está agrandada y engrosada (Figura 4), pero las trabeculaciones generalmente están ausentes.9 Esto contrasta con la vejiga de un paciente con válvulas uretrales posteriores, que suele estar marcadamente trabeculada. Durante la uretrocistografía, la vejiga, por lo tanto, es típicamente de pared lisa y puede mostrar un remanente de uracal o un divertículo. Los remanentes de uraco pueden desplazar la vejiga a la pared abdominal. El 25-50% de los casos presentan quistes de uraco o un uraco completamente permeable36,42,43. El trígono suele agrandarse con orificios ureterales grandes colocados lateralmente, lo que explica la asociación común con el reflujo vesicoureteral9. Por lo general, el trígono desemboca en un cuello vesical bien abierto y ancho.39
Desde el punto de vista histológico, la vejiga parece mostrar dos patrones en pacientes con SPB. En pacientes con obstrucción del tracto urinario inferior, las vejigas engrosadas muestran una mayor cantidad de músculos y una proporción normal de colágeno a músculo. Las vejigas sin obstrucción muestran una pared vesical más delgada con un aumento en la proporción de colágeno a músculo.57 La inervación de la vejiga parece ser normal con una distribución normal de células ganglionares.4
La evaluación urodinámica de la vejiga de pacientes con SPB por lo general revela una vejiga de gran capacidad con presiones de reposo normales.4 Estos pacientes generalmente también muestran una primera sensación de llene tardía con una contractilidad deficiente de la vejiga.58,59 La inestabilidad de la vejiga con contracciones no inhibidas puede ocurrir, pero generalmente son raras .58-60 Kinahan et al informaron en su serie que el 44% de sus pacientes podían vaciar espontáneamente, con presiones de vaciado normales y bajos volúmenes residuales; mientras que los otros requirieron una cateterización intermitente limpia59. Ciertamente, esta pobre contractilidad, así como el reflujo vesicoureteral masivo observado en estos pacientes, darían cuenta de los altos niveles de orina residual que pueden verse en estos niños.
Próstata y uretra posterior
La uretra prostática es típicamente alargada y se estrecha en la uretra membranosa (Figura 5). Además, el cuello de la vejiga suele estar amplio y dilatado. Estas características le dan a la uretra prostática una "forma triangular" típica en las radiografías.33 La pared prostática anterior es típicamente más corta que la pared posterior. El verumontanum suele ser pequeño o ausente y puede haber un divertículo utricular.33 Una verdadera obstrucción a nivel de la uretra prostática o membranosa es rara, y solo ocurre en el 10-20% de los casos.33 Como se mencionó anteriormente, en los pacientes en que se demuestra una obstrucción uretral verdadera, generalmente se consideran la forma más grave de SPB. Sin una verdadera obstrucción uretral, Stephens ha postulado que puede haber una acodadura de la uretra prostática secundaria a las diferencias en la longitud entre la próstata anterior y posterior. Esto, puede entonces dar cuenta de una obstrucción funcional, al crear un flap valvular de la uretra.
El análisis histológico de la próstata en pacientes con SPB, ha revelado una hipoplasia severa, con un mínimo de músculo liso y glándulas tubuloalveolares.38 En comparación, las próstatas de pacientes con válvulas uretrales posteriores revelaron una cantidad normal de músculo liso y parénquima glandular que estaba comprimido, pero por lo demás normal . Esto llevó a Moerman a especular que la próstata dilatada e hipoplásica es la causa, más que el efecto, del SPB.38
Uretra anterior
Por lo general, la uretra anterior en estos pacientes es normal, aunque ha habido informes tanto de atresia uretral como de micro uretras, así como del desarrollo de la megalouretra en estos pacientes. Los pacientes con atresia uretral o con una micro uretra generalmente sobreviven de forma secundaria a un uraco permeable. Algunos han sugerido que estas uretras estrechas se desarrollan normalmente, pero están infrautilizadas y, por lo tanto, pueden manejarse con dilatación progresiva.
Los dos tipos de megalouretra, escafoides y fusiformes, pueden estar asociados con este síndrome. En la variedad fusiforme, los cuerpos cavernosos son deficientes, así como el esponjoso. Esta forma es más grave y puede asociarse con displasia renal y otras anomalías letales.62 La megalouretra escafoides, que es la forma más común y menos grave, se caracteriza solo por una deficiencia del esponjoso, con preservación de los cuerpos cavernosos, el glande y fosa navicularis.33 Se cree que un defecto mesodérmico podría explicar este mal desarrollo uretral.63
Órganos sexuales masculinos accesorios
Se han observado anomalías en el conducto deferente, epidídimo y vesículas seminales. Dada la derivación mesodérmica común de estos órganos, estos hallazgos no son sorprendentes. El epidídimo se suele desprender de los testículos y se ha descubierto que el deferente puede drenar ectópicamente.64 Además, las vesículas seminales pueden estar dilatadas, atréticas o ausentes.42 Todos estos hallazgos, junto con la histología testicular anormal son importantes para la fertilidad futura de estos pacientes.
Testículos
La criptorquidia bilateral se considera un sello distintivo de este síndrome, ya que la mayoría de estos testículos son intraabdominales por encima del nivel de los vasos ilíacos.33 Se sugirió originalmente que los gubernáculos eran normales, aunque alargados y unidos al anillo inguinal interno; sin embargo, Elder y otros han sugerido que el gubernáculo puede ser realmente anormal e incluso ausente.4,45
Histológicamente, los testículos también demuestran una variedad de anomalías. Orvis y otros han informado de una reducción en el número de espermatogonias, así como en la hiperplasia de células de Leydig.65 Además, Massad y otros han demostrado células germinales atípicas con núcleos anormales.66 Estas células germinales anormales se parecen a las neoplasias testiculares intratubulares y, por lo tanto, a los niños con SPB debe vigilarse para detectar el desarrollo de tumores testiculares. De hecho, se han documentado casos de tumores testiculares en estos pacientes, pero en general el riesgo no parece ser mayor que en otros pacientes con testículos no descendidos.67-69
Función sexual y fertilidad
La función sexual en pacientes con SPB parece estar intacta. Siempre que los cuerpos cavernosos sean normales, estos pacientes pueden experimentar erecciones y orgasmos normales y pueden mantener relaciones sexuales satisfactorias70. Sin embargo, dado el cuello vesical abierto, estos pacientes experimentan eyaculación retrógrada y tienden a ser azoospérmicos70,71. No hay informes de padres con SPB con reproducción natural. Hasta la fecha, el único caso reportado de paternidad en un paciente con SPB pasa con la inyección intracitoplásmica de esperma.72 Claramente, la infertilidad asociada con este síndrome es multifactorial ya que existen anomalías histológicas de los testículos y la próstata, así como anomalías anatómicas de la próstata , cuello vesical, vasos deferentes, vesículas seminales y testículos.
Anormalidades extra genitourinarias
Pared abdominal
La característica de la piel arrugada, "parecida a una ciruela" del recién nacido es uno de los hallazgos característicos en niños con SPB (Figura 1). Este aspecto es secundario a la hipoplasia del músculo que se produce en las tres capas musculares de la pared abdominal.40 Los músculos rectos superiores y los músculos oblicuos externos suelen estar mejor desarrollados y el grado de hipoplasia puede ser variable y asimétrico 9, 33. En orden de afectación de mayor a menor de los músculos involucrados es: transversus abdominis, rectus abdominis debajo del ombligo, oblicuo interno, oblicuo externo y rectus abdominis sobre el ombligo.6 El músculo abdominal superior más desarrollado arriba produce un desplazamiento cefálico del ombligo. De hecho, la masa muscular disminuida puede permitirle ver la peristalsis intestinal a través de la piel.
La pared abdominal dañada puede dificultar la capacidad del recién nacido para sentarse desde la posición supina y retrasar la capacidad del niño para caminar.33 De lo contrario, no parece haber ningún otro obstáculo para la actividad física normal. La falta de músculo abdominal también puede resultar en una pobre capacidad de tos y puede contribuir a un aumento de la incidencia de infecciones respiratorias.73 Los músculos abdominales deficientes también pueden contribuir al estreñimiento.39 Sin embargo, en general, parece que, aunque la laxitud de la pared abdominal sea profunda, las limitaciones físicas de esta deficiencia son, en realidad, mínimas.
La evaluación de la pared abdominal ha revelado una distribución normal de los nervios y un aporte sanguíneo a los músculos.40,74 Las fibras musculares aparecen al azar y, en los casos más graves, están totalmente ausentes. Microscópicamente, las fibras pueden reemplazarse por una gruesa capa de colágeno.9 Además, Mininberg et al utilizaron microscopía electrónica para visualizar la desorganización de la banda Z y los agregados de glucógeno.40
Anomalías ortopédicas
Las anormalidades ortopédicas ocurren en el 45-63% de los pacientes con SPB. 75 Las anomalías más frecuentes son dislocación de cadera y talón equinovaro. 51,76 Además, las deformidades de pectus carinatum y pectus excavatum están comúnmente presentes, mientras que la escoliosis es la anomalía espinal más común.76,77 Se ha encontrado que el crecimiento está alterado en el 32% de los pacientes con SPB y se ha relacionado con una función renal deficiente78 El retraso del crecimiento parece ser más profundo en el primer año de vida, pero el crecimiento de recuperación parece permanecer alterado a largo de la vida78.
Anormalidades gastrointestinales
Las anomalías gastrointestinales parecen ocurrir en hasta el 30% de los niños afectados. Las anomalías como la mal rotación, la atresia, la estenosis y el vólvulo parecen ser secundarias a la persistencia del mesenterio embrionario.79 Los mismos defectos en la suspensión también pueden permitir que el bazo se desplace libremente y se ha informado de torsión esplénica.80 Se ha observado gastrosquisis, ano imperfecto y onfaloceles. 79,81-83 También puede ocurrir estreñimiento que puede ser el resultado de una ausencia de tono muscular abdominal. Además, se ha informado asociación con la enfermedad de Hirshsprung, lo que también puede contribuir al estreñimiento. Debido a estas asociaciones gastrointestinales informadas, se recomienda que todos los pacientes con SPB se sometan a una evaluación radiográfica de su tracto gastrointestinal79.
Anomalías cardíacas
Se ha informado que se producen anomalías ** ** cardíacas en el 10% de los pacientes. Los hallazgos cardíacos asociados incluyen defectos del tabique ventricular y atrial, conducto arterioso permeable y tetralogía de Fallot.51 Dadas estas asociaciones, se recomienda evaluación cardiológica en todos los pacientes portadores de SPB.
Anomalías pulmonares
Como el desarrollo pulmonar en el útero depende de cantidades adecuadas de líquido amniótico, los fetos con insuficiencia renal grave también pueden desarrollar hipoplasia pulmonar. En los peores casos, la hipoplasia pulmonar grave es incompatible con la vida y puede causar la muerte en el período perinatal.51 Se estima que el 20% de los recién nacidos morirá en el período perinatal secundario a problemas pulmonares.71,84 De los que sobreviven, el 55% sufrirá problemas pulmonares significativos y prolongados78. La pared abdominal laxa puede afectar la mecánica de ventilación y puede aumentar la incidencia de neumonía.85,86 Estos mismos problemas también pueden contribuir al desarrollo de bronquitis y compromiso respiratorio que puede ocurrir después de la anestesia 78,86
También hay un subconjunto de pacientes con características incompletas de este síndrome y se han denominado "pseudoprune". A menudo, estos individuos no tendrán los hallazgos característicos de la pared abdominal, pero sufrirán de criptorquidia bilateral y también demostrarán los hallazgos asociados del tracto urinario.87 Estos pacientes todavía tienen riesgo de desarrollar insuficiencia renal y, por lo tanto, deben seguirse de cerca.87
PBS en niñas
El cinco por ciento de los niños con SPB son mujeres. Las anomalías asociadas en estos niños incluyen la apariencia típica de la pared abdominal y las anomalías del tracto urinario (Figura 6) .11 Otros hallazgos en estas pacientes incluyeron una alta tasa de atresia vaginal, duplicación uterina y ano imperforado. Reinberg et al, en su serie de mujeres con SPB, también demostraron una alta tasa de hipoplasia y displasia renal, similar a sus contrapartes masculinas. En estas niñas se presentaron con frecuencia atresia uretral, duplicación uterina y anomalías ano rectales. La tasa de mortalidad perinatal fue alta; de los cuatro pacientes sobrevivientes, la insuficiencia renal se desarrolló en dos requiriendo trasplante renal.
PRESENTACIÓN Y EVALUACIÓN
Diagnóstico prenatal e intervención
El diagnóstico de SPB se puede realizar desde las 11 semanas de gestación mediante ecografía prenatal88. Los hallazgos ecográficos asociados incluyen oligohidroamnios o anhidramnios, hidrouréter, vejiga distendida y una pared abdominal delgada y atenuada. Síndrome de megacistis megaureter o válvulas uretrales posteriores, pueden tener hallazgos similares en la ecografía fetal. Aunque las imágenes de ultrasonido modernas han mejorado, aún puede ser difícil establecer un diagnóstico prenatal definitivo. Informes anteriores han demostrado que la precisión diagnóstica prenatal para determinar la etiología de la hidronefrosis varía entre el 30 y el 85% .90
La identificación de los hallazgos prenatales anteriores ha llevado a algunos a recomendar la colocación intrauterina de una derivación vesicoamniótica para descomprimir el tracto urinario y establecer una cantidad normal de líquido amniótico o sugerir la interrupción del embarazo.91-94 Sin embargo, la dilatación no siempre representa la gravedad de la obstrucción ni refleja la función renal postnatal. Por lo tanto, puede ser difícil justificar la necesidad de cualquier intervención o terminación del embarazo utilizando solo el grado de hidronefrosis95. Los criterios utilizados para la derivación intrauterina incluyen un cariotipo normal, oligohidroamnios severos y una función renal fetal normal94. La decisión de intervenir se basa en evaluaciones seriadas de la química fetal de la orina como el sodio, el cloro, la osmolaridad, la microglobulina y las proteínas totales. Los fetos con valores altos de osmolaridad y sodio en la orina tienen el peor pronóstico. En el raro caso de oligohidramnios progresivos con atresia uretral97 u obstrucción durante el trabajo de parto debido a una vejiga distendida masivamente la intervención antenatal puede ser beneficiosa91. A pesar de varios informes de casos que documentan la capacidad para realizar este procedimiento, las revisiones no han demostrado una mejoría sostenible en la insuficiencia renal y de la función pulmonar.90,98,99 Además, se pueden producir complicaciones en la colocación de la derivación, incluida la gastrosquisis traumática secundaria a un defecto de la pared abdominal.100 Freedman et al también demostraron una tasa de mortalidad del 38% y una tasa de insuficiencia renal del 29% en niños que se sometieron a intervenciones intrauterinas.97 Parecería que la intervención intrauterina tiene cierto potencial, pero aún no alcanza el beneficio deseado de mejoría renal o pulmonar. Dadas estas circunstancias, la intervención fetal requiere una evaluación cuidadosa de riesgo / beneficio y una discusión exhaustiva con los padres del niño.
Atención peri parto
Debido a que el recién nacido puede requerir asistencia ventilatoria y atención neonatal avanzada, es prudente tener el parto donde se encuentren disponibles dichas atenciones neonatales. El parto puede complicarse con parto prematuro, corioamnionitis o el atrapamiento fetal debido a la megavejiga. La hipoplasia pulmonar asociada en los bebés con SPB puede conducir a neumotórax. Para los recién nacidos que tienen hipoplasia pulmonar, se requerirá asistencia avanzada, como ventilación de alta frecuencia y, posiblemente, óxido nítrico inhalado (iNO) .101 Un equipo obstétrico y neonatal con experiencia debe estar presente en el momento del parto.
Período neonatal
El aspecto característico del abdomen en forma de ciruela generalmente sugiere el diagnóstico (Figura 1). Se requiere un enfoque multidisciplinario para la evaluación inicial, específicamente la participación del neonatólogo, nefrólogo y urólogo. La participación de otras disciplinas dependerá del perfil clínico. Si hay compromiso cardíaco o pulmonar, la evaluación y el tratamiento de estos tienen prioridad durante el examen post natal inmediato. La evaluación genitourinaria se puede realizar una vez que el niño esté estable.
Evaluación de la función renal
Una creatinina inicial sirve como una línea de base, pero el valor estará relacionado con la creatinina materna los primeros días de vida. Las mediciones de creatinina sérica en serie son esenciales para determinar la función renal neonatal y para controlar la tendencia. Un aumento en la creatinina durante las primeras semanas augura un mal pronóstico. Si se observa que la creatinina tiene un nadir de menos de 0.7 ng / dl, se ha observado que la insuficiencia renal posterior es menos probable.102 Además, los electrolitos séricos también deben ser monitoreados y pueden ser útiles para evaluar la función renal en general.
Imágenes
La evaluación radiográfica del recién nacido debe comenzar con la radiografía de tórax. Esto ayudará a descartar cualquier complicación pulmonar asociada como resultado de la oligohidramnios que se puede observar con el PBS, específicamente el neumotórax, el neumomediastino y la hipoplasia pulmonar.103 La ecografía renal y vesical puede evaluar el parénquima, la presencia de quistes corticales y distensión vesical. Se debe obtener un cistouretrograma de evacuación (VCUG) (Figuras 3, 5) para diferenciar entre obstrucción y estasis. Idealmente, los antibióticos profilácticos deben iniciarse antes de obtener un VCUG para minimizar el riesgo de infección. En el recién nacido con función renal normal y evidencia clínica de vaciamiento vesical, la VCUG puede retrasarse, ya que casi el 70% de los niños tiene reflujo vesicoureteral. 105, 106 La exploración renal de medicina nuclear con 99mTcDMSA o 99mTcMAG3 ayuda a identificar el tejido renal en funcionamiento. Sin embargo, la estasis debida a la dilatación ureteral y la tortuosidad puede impedir cualquier diagnóstico de obstrucción con la renografía con Lasix (Figura 7). La renografía se puede obtener después de 4 semanas para permitir cambios durante la fisiología neonatal.
MANEJO
Espectro de PBS
Dado que existe un amplio espectro de características de presentación en pacientes con SBP, Woodard describió tres categorías principales de presentación en el período neonatal.107 Aunque no existe una delineación clara entre las categorías, esta clasificación ayuda a planificar la evaluación y el manejo del recién nacido.
Categoría 1
La displasia renal u obstrucción uretral secundaria a atresia que da como resultado oligohidramnios marcado. Esto a su vez resulta en hipoplasia pulmonar severa. Puede haber una constelación de otros sistemas involucrados. La hipoplasia pulmonar y/o la insuficiencia renal suelen causar la muerte en el período neonatal. Dado que estos neonatos tienen otros problemas que amenazan la vida, sólo se indica una intervención urológica mínima y a menudo se limita a la cateterización de la vejiga.
Categoría 2
Tienen un grado variable de insuficiencia renal, dilatación del tracto superior y reflujo vesicoureteral. Como rara vez tienen insuficiencia pulmonar o renal temprana potencialmente mortal, la evaluación y el manejo del tracto urinario asumen importancia. Requieren un enfoque individualizado debido a los diversos grados de presentación. El tratamiento específico del tracto urinario dilatado en este grupo ha sido controvertido durante los últimos 25 años.108, 109 Existe una superposición entre las características clínicas de los pacientes de Categoría II y Categoría III y diferentes proporciones de estos se incluyen en las series informadas. Además, el seguimiento a largo plazo es limitado. Por lo tanto, es difícil comparar los resultados de varias series que abogan por un manejo quirúrgico agresivo o un enfoque conservador. Los pacientes en la categoría 2 requieren un enfoque individualizado debido a los distintos tipos de presentación. Dado que la infección urinaria y la insuficiencia renal progresiva son la principal causa de morbilidad y mortalidad en este grupo, se han realizado terapias quirúrgicas agresivas y, como resultado, se ha observado una mejoría funcional y anatómica.84,108,110 El mejoramiento del resultado renal y la disminución de las infecciones del tracto urinario se han documentado con el "tailoring" ureteral , acortamiento y reimplante, minimizando así la estasis y el reflujo.84,110 Otros han abogado por la observación cercana de estos pacientes con monitoreo de infección del tracto urinario e intervención quirúrgica para aquellos con obstrucción del tracto urinario o infección bacteriana recurrente.109 Woodhouse et al, en una serie de 47 casos, tuvieron 13 neonatos que presentaron infección y dilatación del tracto urinario. En 12 se realizó derivación urinaria alta. Se intentó una reconstrucción adicional del tracto superior en seis de estos casos, pero la micción normal y la función renal estable solo se lograron en tres.111 Sin embargo, debido a la amplia variación de las características de presentación en esta población de pacientes y los limitados datos publicados, no se puede hacer una recomendación general con respecto a los pacientes que entran en esta categoría. El plan de tratamiento debe ser hecho a medida según las comorbilidades, la función renal, las infecciones, el cuadro clínico general y las preferencias de la familia
Categoría 3
Este grupo está compuesto por niños con la tríada de características incompletas o leves, uropatía leve a moderada, sin displasia renal, función renal estable y sin hipoplasia pulmonar. Su función renal suele ser conservada. Requieren monitorización regular con ultrasonido y creatinina. A menos que haya un empeoramiento de la función renal o una ITU recurrente, no requieren ninguna cirugía del tracto urinario. En ausencia de indicaciones para la reconstrucción del tracto urinario, la orquidopexia bilateral se lleva a cabo en la infancia. Cualquier deterioro en la función renal o nueva infección del tracto urinario requiere una evaluación adicional. La urodinamia puede ser necesaria para evaluar la vejiga y la idoneidad de la evacuación.58
Tratamiento quirúrgico
Derivación urinaria
Puede ser necesaria durante el tratamiento inicial, en vista de la uro sepsis, la obstrucción del tracto urinario o el empeoramiento de la función renal. Si la micción es ineficiente debido a la estenosis uretral, válvulas o atresia, se requerirá una derivación urinaria. En los recién nacidos con obstrucción al tracto de salida de la vejiga puede ser que el uraco está patente, pero como se cierra en las primeras semanas de vida, el drenaje a través de este sea impredecible.
Vesicostomía
Cuando está indicado, la vesicostomía cutánea es un método rápido y eficiente para desviar la orina al nivel de la vejiga y comúnmente se emplea la técnica de Blocksom. Es importante construir el estoma utilizando la cúpula de la vejiga. Como la vejiga es grande y flexible, si la cistotomía se realiza más abajo en la pared anterior de la vejiga, es propenso a prolapso y obstrucción. Se recomienda un estoma más grande ya que la estenosis del estoma es común. Un uraco permeable o un divertículo grande en el uraco se encuentran con frecuencia. Si está presente, el divertículo o el uraco permeable pueden extirparse en el momento de la vesicostomía.
Derivación supravesical
En la mayoría de los casos, la vesicostomía es adecuada para la descompresión efectiva del sistema urinario. En el contexto de reflujo masivo, una ureterostomía refluyente en asa unilateral puede permitir la descompresión adecuada de ambos riñones y, al mismo tiempo, permitir el ciclo normal de la vejiga. Sin embargo, en el contexto de la obstrucción en la unión ureterovesical o ureteropélvica, se recomienda la derivación urinaria proximal. Si se anticipa una reconstrucción ureterovesical, es preferible evitar una ureterostomía alta, ya que el uréter superior se utiliza para la reconstrucción y la vascularización de este segmento puede verse comprometida por la ureterostomía.56,113 Una ureterostomía en asa baja puede descomprimir adecuadamente tanto el riñón ipsilateral como la vejiga sin poner en peligro la reconstrucción posterior. La ureterostomía cutánea terminal puede considerarse si no se sospecha obstrucción ureteral superior. Además de aliviar la obstrucción, la anatomía uretérica superior no se altera lo que es deseable para una futura reconstrucción.113 Otra alternativa para el drenaje del tracto superior es la pielostomía cutánea. Los bebés que requieren una derivación temporal que requiera una reconstrucción completa planeada en una fecha posterior pueden beneficiarse de una pielostomía inicial seguida de cistoplastia de reducción, orquidopexia bilateral y abdominoplastía una vez que están más grandes.114 Sin embargo, la "reconstrucción total" tiene poco papel en el manejo moderno del SPB y rara vez se indicará una pielostomía. Cuando el paciente se encuentra séptico y no está en condiciones de recibir anestesia general, puede indicarse realizar nefrostomías percutáneas.
Reconstrucción
La reconstrucción correspondiente al sistema urogenital incluye la orquidopexia, abdominoplastía y reconstrucción del tracto urinario. Los testículos no descendidos son un sello distintivo del Síndrome de Prune Belly y existe un acuerdo universal con respecto a la necesidad de orquidopexia. El defecto de la pared abdominal puede variar desde una leve divergencia de los músculos rectos abdominales hasta una deficiencia severa de la pared abdominal. La decisión de la abdominoplastía depende en gran medida de la extensión de la deformidad y la cosmética deseada. Por otra parte, la mejora en el vaciado de la vejiga es una consideración adicional para ofrecer abdominoplastía en el SPB.115 La uropatía obstructiva verdadera, las infecciones urinarias recurrentes y la insuficiencia renal crónica en anticipación de la terapia de reemplazo renal son indicaciones relativas para la intervención quirúrgica. No está claro si la corrección del reflujo de alto grado y / o la restauración del calibre y la capacidad normal del tracto urinario tienen algún efecto para minimizar el deterioro renal o reducir el riesgo de infecciones y existe una controversia considerable con respecto a las indicaciones y el momento de la reconstrucción del tracto urinario. Para mejorar la función del detrusor, se ha sugerido la cistoplastia de reducción116, pero esto no parece disminuir la capacidad de la vejiga ni mejorar la dinámica de evacuación a largo plazo.117 Como no hay criterios definidos para asignar las categorías y por ende clasificar adecuadamente los pacientes con SPB, es difícil comparar los resultados de diferentes series.
Uretra
El espectro de anomalías uretrales incluyen: atresia uretral o microuretra, megalouretra u obstrucción en la uretra prostato-membranosa. Se ha sugerido la dilatación uretral sucesiva según lo descrito por Passerini-Glazel et al usando la técnica PADUA para la atresia uretral, pero los resultados no han sido uniformemente exitosos61. Si existe una vesicostomía, es posible realizar dilataciones anterógradas y retrógradas. En una serie informada por González et al, 4 de 6 niños algún tipo de derivación supravesical después de múltiples intentos fallidos de dilatación.118 También puede requerirse uretrostomía perineal inicial antes de la reconstrucción.119
Como se mencionó anteriormente, la megalouretra puede ser de tipo fusiforme o escafoides. La megalouretra escafoides es más común. La técnica de reparación fue descrita inicialmente por Nesbit en 1955. Después de realizar una incisión circuncidante y deglobamiento del pene, se realiza una uretroplastia de reducción longitudinal sobre un catéter. Esta técnica es sencilla, evita las líneas de sutura superpuestas y sus principios se mantienen en uso hoy en día.120 Una técnica alternativa es la plicatura de la uretra, como describen Heaton y sus colegas.121 La reconstrucción puede ser técnicamente desafiante en la variante fusiforme, ya que no existe esponjoso y la uretra a menudo se dilata en forma masiva. Las opciones son limitadas, debido a la falta de tejido periuretral. El falo generalmente está muy agrandado y puede tener una capacidad incluso mayor que la vejiga nativa. Sin embargo, ha habido informes de reconstrucción fálica en estos pacientes con resultados estéticos y funcionales satisfactorios122. La rareza del defecto impide cualquier generalización con respecto al tratamiento quirúrgico; cada caso debe ser considerado individualmente.
Dado que la uretra prostática es hipoplásica, hay un cambio abrupto en el calibre en la unión prostatomembranosa, pero la obstrucción verdadera es infrecuente. Ocasionalmente, hay obstrucción de la uretra prostato-membranosa, clasificada por Stephens como una válvula de tipo IV. Está causada por tejido redundante en la uretra membranosa y se puede realizar una incisión transuretral.123 Woodhouse et al siguieron a 29 pacientes en la Categoría III que estaban bien al nacer, con función renal estable. Cuando hubo un deterioro del tracto urinario superior, se observó que la obstrucción se encontraba generalmente en la uretra, lo que se resolvió endoscópicamente mediante la resección del anillo estrecho o por una uretrotomía longitudinal completa.111 En niños con SPB con residuos post-miccionales altos y/o hidroureteronefrosis o RVU en aumento , la uretrotomía interna se puede considerar, aunque no se ha demostrado un éxito a largo plazo124.
Vejiga
La evaluación urodinámica está indicada en el contexto de infecciones del tracto urinario y/o volúmenes post miccionales elevados.58 Se ha postulado que la presencia de resistencia en la unión vesicouretral, junto con la contractilidad vesical deficiente y el reflujo vesicoureteral de alto grado, contribuyen al aumento del residuo post miccional. En los niños que no pueden mantener volúmenes residuales bajos, algunos autores han propuesto una uretrotomía interna para disminuir la resistencia del esfínter y permitir un mejor vaciado de la vejiga. 58, 109 Además, el cateterismo limpio intermitente puede facilitar un mejor vaciado de la vejiga.
Cistoplastia de reducción
Una mala función vesical, junto con una capacidad aumentada y el reflujo vesicoureteral masivo, pueden llevar a una disminución del vaciamiento vesical. Con el fin de mejorar el vaciado de la vejiga, Perlmutter propuso inicialmente la cistoplastia de reducción como un medio para reconstruir la forma de la vejiga a una apariencia más esférica.116 Sin embargo, al largo plazo, el volumen de la vejiga regresó a la línea de base o aumentó en comparación con los volúmenes de cistoplastia de reducción previa.117 No se observan diferencias en la urodinamia en pacientes con reconstrucción versus los que no se realizó.59 Por lo tanto, la cistoplastia de reducción se reserva para la extirpación de un segmento de uraco dilatado que puede verse en asociación con anomalías uretrales, o para extirpar un gran divertículo de uraco.
Reconstrucción del tracto urinario superior
El reflujo vesicoureteral ocurre en aproximadamente el 70% de los niños con SPB. El enfoque conservador en estos pacientes consiste en la profilaxis antibiótica y el monitoreo seriado de la función renal con objetivos de mantener la función renal y evitar la cirugía.113 Se observó que la derivación proximal inicial con reconstrucción posterior arrojó resultados que son igualmente exitosos y comparables con la cirugía reconstructiva primaria realizada en la infancia temprana o en la infancia tardía.115 En el contexto de pielonefritis recurrente o el deterioro renal progresivo, se recomienda "tailoring", enderezamiento y la reimplantación ureteral después de extirpar el uréter dilatado redundante (especialmente su tercio distal). Esto se puede lograr mediante una plicatura o una remoción, con igual éxito. 106 Sin embrago, la tunelización de la submucosa puede ser más desafiante si se realizaron plicaturas debido al mayor volumen ureteral resultante. En ocasiones, el uréter distal tortuoso redundante se puede extirpar con una reimplantación estándar del uréter proximal de tamaño más normal.
Orquidopexia
Los testículos no descendidos son una de las características de este síndrome. El momento de la orquidopexia depende si el paciente está apto para una anestesia y la necesidad de abdominoplastía concomitante o reconstrucción del tracto urinario. Como la función endocrina está conservada y existe un potencial maligno, es deseable una orquidopexia temprana. La mayoría de estos testículos son intraabdominales, altos en la pared pélvica posterior que recubren los vasos ilíacos y, por lo tanto, es improbable el descenso espontáneo. La orquidopexia estándar, sin división de los vasos testiculares, puede ser más exitosa si la orquidopexia se realiza durante la infancia temprana.125 Dado que los testículos se ubican en el abdomen a nivel de los vasos ilíacos, la movilización proximal adecuada es difícil con el abordaje inguinal estándar y se prefiere el abordaje transabdominal. Si se realiza reconstrucción urinaria o abdominoplastía concomitante, los vasos pueden movilizarse hasta el origen para asegurar una movilización adecuada para la orquidopexia en una sola etapa. El objetivo principal de la orquidopexia es preservar la función hormonal y permitir un examen fácil, ya que se ha documentado el potencial maligno66. Además, aunque el potencial de fertilidad está comprometido, las técnicas de reproducción asistida podrían ser posibles. Hay un reporte de espermatozoides maduros en la orina postcoital en un paciente que se sometió a una orquidopexia neonatal.115 Se encontró que los niveles de testosterona eran más altos en los niños que se sometieron a una orquidopexia en la infancia temprana en comparación con aquellos que habían organizado la orquidopexia de Fowler-Stephens en una edad mayor. Por lo tanto, se ha recomendado la orquidopexia precoz, desde los seis meses. 115 Si no se indica un procedimiento urológico adicional y no se planifica la abdominoplastía, la orquidopexia laparoscópica es una opción a considerar. La disección puede llevarse a cabo hasta el origen de los vasos testiculares, facilitando así la orquidopexia en una sola etapa. La anatomía alterada de estos niños presenta desafío para el procedimiento laparoscópico. Dado que el uréter y los riñones a menudo están muy dilatados, se debe tener cuidado durante la disección.126 La técnica laparoscópica de Fowler-Stephen se ejecuta como un abordaje de una etapa o dos etapas con un éxito razonable cuando no es posible realizar una orquidopexia en una sola etapa.126,127 El autotrasplante microvascular una alternativa para los testículos intraabdominales muy altos que no se pueden bajar al escroto a pesar de la movilización adecuada.128
Seguimiento a largo plazo de la orquidopexia
En paciente entre 16 y 28 años en que se efectuaron orquidopexias en la infancia tardía o la adolescencia, se han reportado erecciones y orgasmos normales, con un porcentaje importante de eyaculación retrógrada. Aunque infértiles, estos hombres eran sexualmente activos.70 La orquidopexia se puede realizar con un abordaje abierto o laparoscópico si se realiza dentro de los primeros años de vida. A menudo, los testículos se pueden bajar al escroto sin un enfoque de Fowler-Stephen. 110 La división de los vasos gonadales no parece afectar negativamente el resultado127. En una serie de 32 niños con SPB que se sometieron a una orquidopexia bilateral, 26 no requirieron la ligadura de las arterias testiculares.
Reconstrucción de la pared abdominal
Al igual que en el tracto urinario, el espectro del defecto de la pared abdominal se extiende desde casos leves hasta la laxitud severa. En aquellos con deficiencia mínima, puede existir mejoría en la medida que el paciente crece y en muchas ocasiones no se requiere intervención. Sin embargo, la mayoría de los niños con SPB de Categoría II tienen un defecto significativo en la pared abdominal. El defecto de la pared abdominal puede no tener consecuencias potencialmente mortales, pero se cree que la ineficiente contracción de los músculos de la pared abdominal afecta la vejiga, el intestino y la función pulmonar.129 Por consiguiente, aunque no hay evidencia suficiente, la reparación de la pared abdominal puede mejorar la tos y la defecación.115 Si se anticipa una reconstrucción del tracto urinario concomitante, es prudente esperar hasta obtener imágenes claras del tracto urinario y de la función de vaciamiento antes de planificar la reconstrucción. Si no se prevé una reconstrucción del tracto urinario, puede combinarse con la orquidopexia durante la infancia. La abdominoplastía junto con la orquidopexia a los 6 meses permite la movilización adecuada de los testículos. Dado que estos bebés tienen hipoplasia pulmonar subyacente101 y la respiración puede verse comprometida aún más por la abdominoplastía, el beneficio en términos de orquidopexia y reconstrucción exitosas debe siempre compararse con el riesgo anestésico, ya que la mortalidad por problemas pulmonares se ha documentado en el post operatorio de una reparación muy precoz.115
Técnica de Randolph
Randolph et al 130 describieron inicialmente la reconstrucción de la pared abdominal basándose en datos electromiográficos (EMG). La cartografía EMG indicó que el área más gravemente afectada es la región infraumbilical. Por lo tanto, recomendaron una incisión transversal desde la punta de la 12ª costilla sobre la espina ilíaca superior anterior hasta la sínfisis púbica y formando una elipse hasta la espina ilíaca superior anterior contralateral y la punta de la 12ª costilla. Se realiza una incisión paralela con extirpación total del exceso de piel, músculo subyacente y peritoneo. La reaproximación de espesor total del margen inferior sano de la fascia, incluido el periostio, se realiza en las espinas ilíacas superiores anteriores y en la sínfisis púbica con sutura no absorbible. La fascia se cierra con sutura interrumpida seguida de una nueva aproximación de la piel. Una serie de 16 pacientes tratados con esta técnica se observó en 7 pacientes abultamiento lateral de la pared abdominal.106
Técnica de Ehrlich
Ehrlich describió su metodología por primera vez en 1986 y luego la modificó en 1993.131,132 La técnica consiste en una incisión vertical con preservación del ombligo. Existe una amplia movilización de la capa muscular lateral de la piel supra yacente y del tejido subcutáneo, seguida de un cierre en "chaleco". Este cierre lleva la capa musculofascial lateral no afectada a la línea media y permite eliminar el exceso de piel y preservar el ombligo.
Técnica de Monfort
La técnica de Monfort (Figura 8) también utiliza una incisión vertical.133 Utilizando una incisión elíptica desde el xifoides hasta la sínfisis púbica y una segunda incisión alrededor del ombligo para su preservación, las capas subcutáneas subyacentes se diseccionan desde la fascia hacia la línea axilar anterior. Se realiza una incisión vertical lateral a las arterias epigástricas superficiales. Se incide el peritoneo parietal posterior. El colgajo muscular en isla central se fija al peritoneo parietal posterior. Los colgajos musculares laterales movilizados se colocan sobre el colgajo muscular central y se vuelven a aproximar en la línea media, de manera similar a una técnica de chaleco. Esta técnica aumenta el grosor de la pared abdominal anterior, crea una cintura y minimiza los abultamientos laterales. Dado que la piel peri umbilical en la línea media no se corta en la técnica de Montfort, el ombligo a menudo aparece demasiado alto después de la reparación. Se ha sugerido una reconstrucción neo-umbilical junto con la abdominoplastía de Montfort.135 Una isla de piel con forma de almendra se conserva a nivel de la cresta ilíaca en la línea media, unida a la dermis. Esto se invagina colocando suturas absorbibles desde debajo de la fascia. Furness et al.136 ha descrito otra modificación de esta técnica. En este enfoque, no se ingresa a la cavidad peritoneal y la disección de la pared abdominal es mínima. Si la orquidopexia ya se había realizado, esta técnica es útil para evitar la entrada en la cavidad peritoneal. 134
Técnica Firlit
En esta técnica, la laxitud de la pared abdominal se evalúa sujetando la línea media con clips y levantando la pared abdominal hasta que esté tensa. Luego, se realiza una incisión en la piel elíptica alrededor de la base de la piel redundante desde el xifoides hasta el pubis, preservando una isla de la piel del ombligo. El exceso de piel se escinde y la laxitud de la placa fascial se corrige con una técnica de plegado con pliegues verticales de espesor total. No se ingresa a la cavidad peritoneal, por lo tanto, este procedimiento es una opción si no se requieren procedimientos intraperitoneales concomitantes. Los autores informaron resultados satisfactorios en 13 pacientes que se sometieron al procedimiento.136
Reconstrucción asistida por laparoscopia de la pared abdominal
Se ha descrito que la reparación asistida por laparoscopía minimiza la entrada a la cavidad peritoneal.137 La cavidad peritoneal se visualiza utilizando un laparoscopio introducido a través de un trocar subxifoideo de 5 mm y la plicatura de la fascia abdominal se realiza mediante la técnica de Firlit 136 Además de la visualización de la visualización de los órganos intra abdominales, cuando se toman las suturas de plicatura profunda, el neumoperitoneo le da al cirujano una idea representativa de cómo se verá el abdomen reconstruido cuando el paciente este de pie, lo que permite una reducción óptima de la pared abdominal.
Resultado de la abdominoplastía
Smith y todos evaluaron los cambios en la micción en doce pacientes que se sometieron a una abdominoplastía de Monfort. Los cambios subjetivos que se produjeron después de la abdominoplastía incluyeron resolución o disminución de la necesidad de doble micción en 9 pacientes, mejoría en la continencia urinaria en 7, mejoría la sensación de llene vesical en 11, mejoría en el flujo urinario en 10 y mejoría para la defecación en 5. También notaron que la incidencia de infecciones del tracto urinario disminuyó de un promedio preoperatorio de 5.7 por paciente por año a 1.2 por paciente por año después de la operación. Además, los volúmenes residuales después de la micción disminuyeron significativamente desde un promedio preoperatorio de 40% a un 13% después de la abdominoplastía, incluso en aquellos en que no se sometieron a una reconstrucción urinaria concomitante, lo que sugiere la influencia de la abdominoplastía para mejorar la micción.129 En la serie por Denes et al, la flacidez mejoró en 29 de los 30 pacientes que se sometieron a una reparación integral. La abdominoplastía mejoró no solo la imagen corporal y la autoestima, sino también la fuerza abdominal, con buenos resultados en el 93,5% de los pacientes. La postura corporal vertical también mejoró en la mayoría de los pacientes. Aunque es difícil evaluar objetivamente la mejora cosmética, esto se comprobó por la satisfacción del paciente y de los padres.112
Trasplante renal
El deterioro de la función renal debido a la displasia renal y/o la pielonefritis recurrente son los motivos de la terapia de reemplazo renal. La incidencia de trasplante renal en pacientes con SPB es de alrededor del 30%. 71 La supervivencia del injerto después del trasplante renal en SPB es similar a los controles de la misma edad, con una supervivencia del injerto de 75% versus 69% en los controles. 138 En comparación con los controles emparejados por edad 10 años después del trasplante, la mejoría en la supervivencia del injerto renal se asoció con la reconstrucción urinaria previa al trasplante para reducir la posibilidad de ectasia e infección urinaria, así como el mantenimiento de bajos residuos post miccionales con un programa de micción por horario y/o de cateterización limpia intermitente. 139 La mayoría de los trasplantes se han realizado sin una abdominoplastía previa. Cuando se encontraron grandes residuos urinarios y ITU después del trasplante, estos mejoraron luego de una abdominoplastía 144. Otra posible complicación es torsión del injerto dado la relativa movilidad libre que presenta el riñón trasplantado en la cavidad abdominal. Esta complicación se puede prevenir con la abdominoplastía y/o nefropexia al momento del trasplante141.
Perspectivas a largo plazo
Debido a la rareza del SPB, la mayoría de la literatura descrita se basa en series de casos. El resultado de los recién nacidos de Categoría I, nacidos con displasia pulmonar, sigue siendo pobre. La mortalidad intrahospitalaria de pacientes recién nacidos con SPB que fallecieron durante su estadía hospitalaria inicial es del 31%. La mortalidad y los trastornos pulmonares se asociaron mucho con la mortalidad hospitalaria.10 Gracias a una mejor atención neonatal y un manejo eficiente del tracto urinario, la supervivencia ha mejorado durante los últimos años. Con el plan de tratamiento y la atención individualizados, la perspectiva general para el paciente de SPB, tanto para la supervivencia como para la calidad de vida, ha mejorado considerablemente, en gran medida gracias a los avances en la atención neonatal, las técnicas quirúrgicas y la evaluación diagnóstica. Aunque el abordaje quirúrgico agresivo puede no ser necesario en todos los pacientes, el seguimiento meticuloso y el manejo activo de la pielonefritis y la obstrucción del tracto urinario son esenciales. Un tercio de las personas con insuficiencia renal en la evaluación inicial desarrollan insuficiencia renal crónica terminal durante la infancia o la adolescencia. El resultado del trasplante renal parece comparable a los pacientes sin SPB de edades similares. La dinámica de la vejiga y el patrón de micción también pueden cambiar con el tiempo. Por lo tanto, el seguimiento a largo plazo es esencial para garantizar una calidad de vida casi normal para los pacientes con el síndrome de Prune Belly.
Figuras
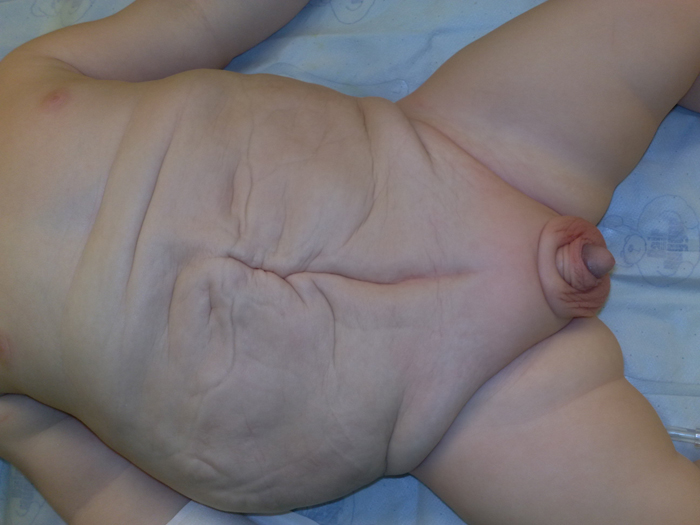
Figura 1: Pared abdominal laxa en paciente con Síndrome de Prune Belly
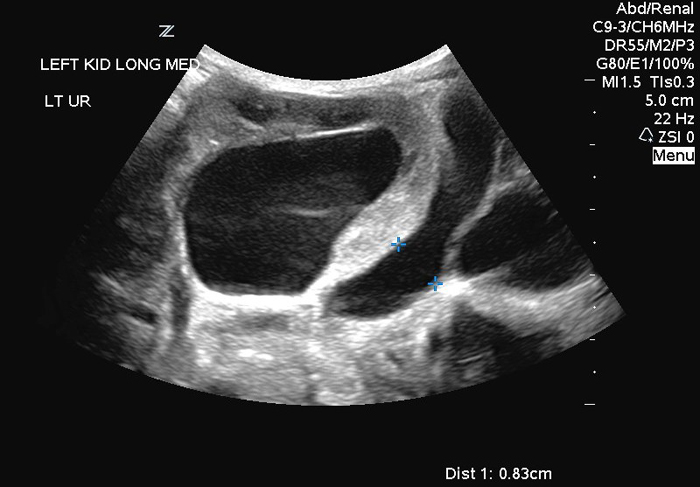
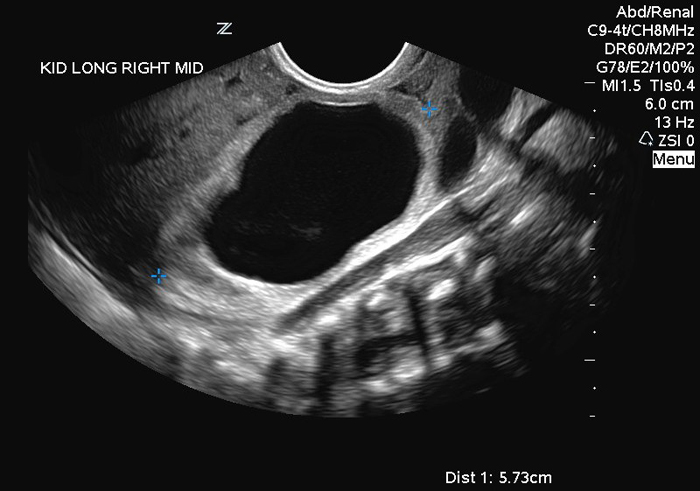
Figuras 2 y 3: Ecografía renal de recién nacido con PBS con hidroureteronefrosis marcada (Fig. 2 A: riñón izquierdo; Fig. 2B: riñón derecho)
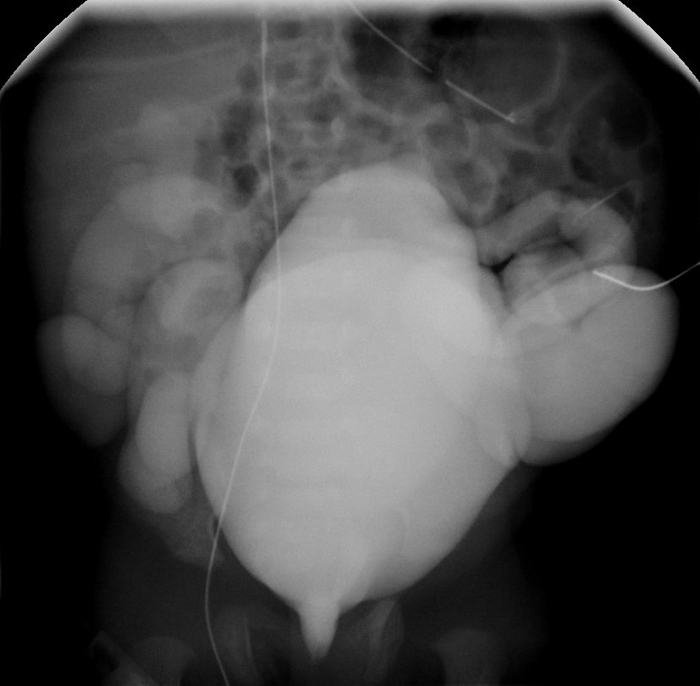
Figura 4: VCUG que muestra un reflujo vesicoureteral bilateral con uréteres dilatados
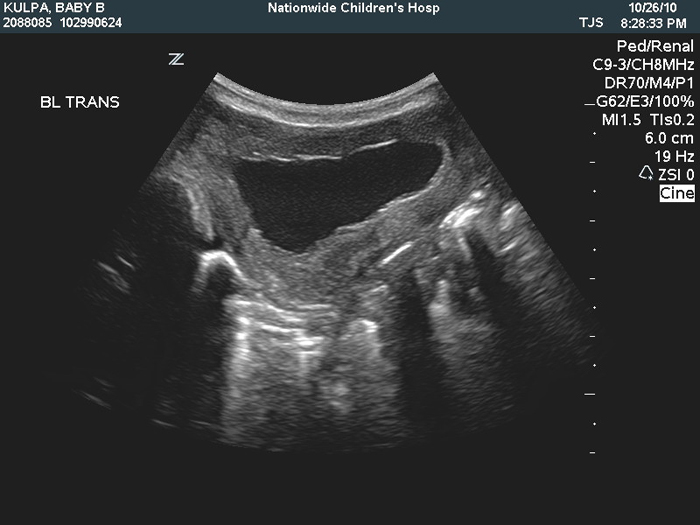
Figura 5: Ecografía de la vejiga que muestra engrosamiento de la pared de la vejiga
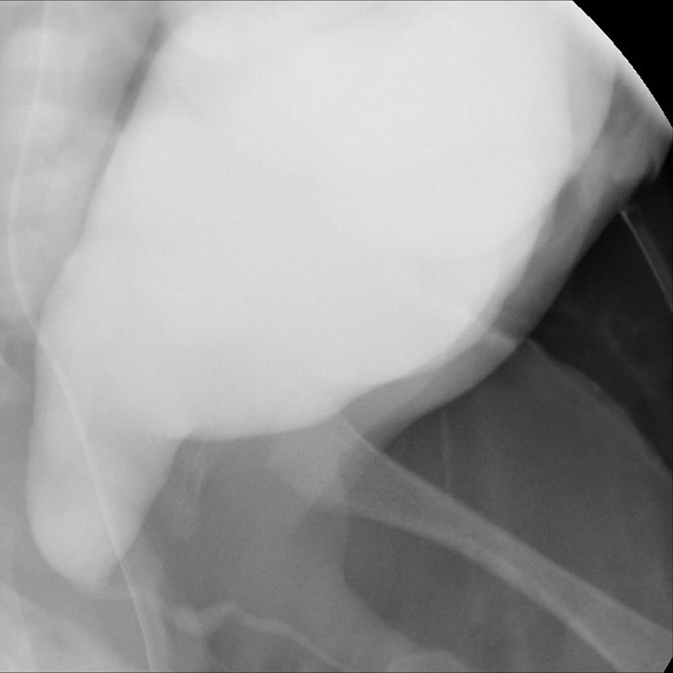
Figura 6: próstata hipoplásica con uretra prostática dilatada
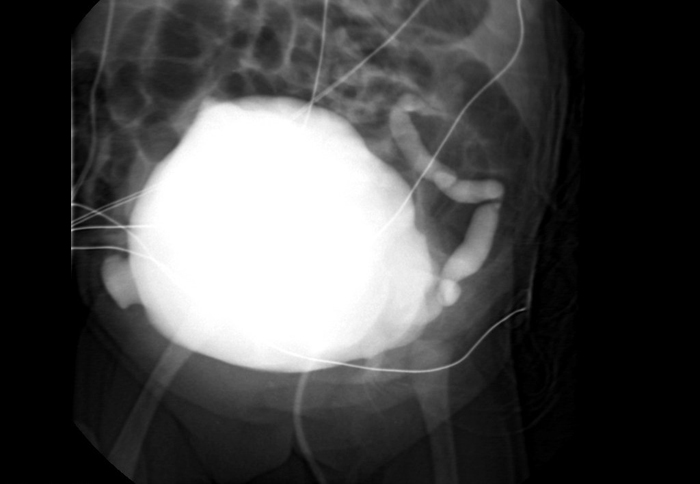
Figura 7: cistograma que muestra la vejiga grande en una niña con PBS
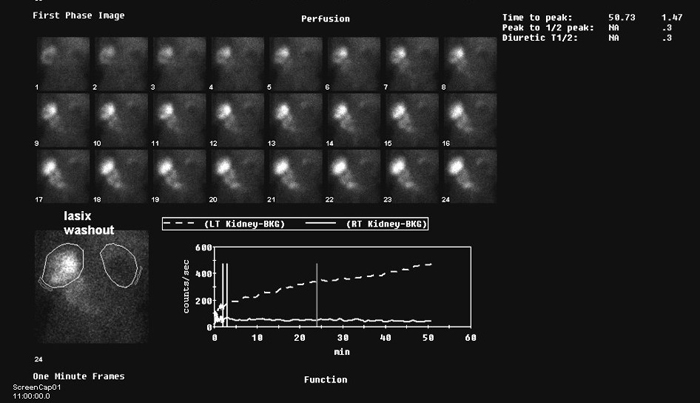
Figura 8: renograma de MAG 2 en un niño con PBS, riñón derecho que funciona mal y dilatación a izquierda con estasis del trazador.
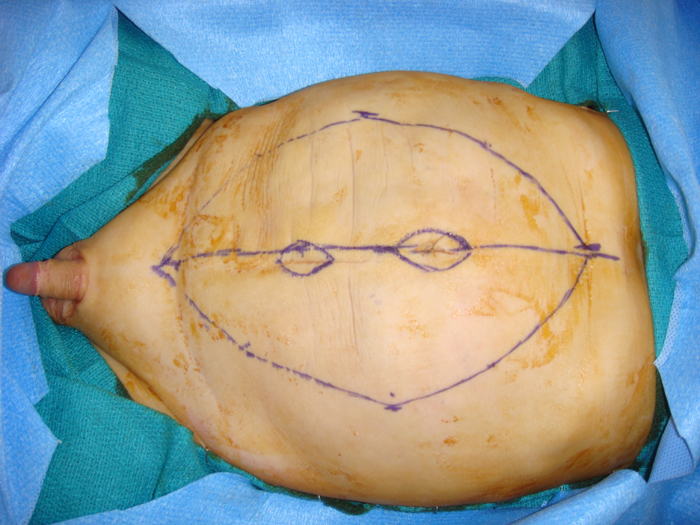
Figura 9: Incisión marcada para abdominoplastía de Montfort. La isla de la piel para neoumbilicus también está marcada.
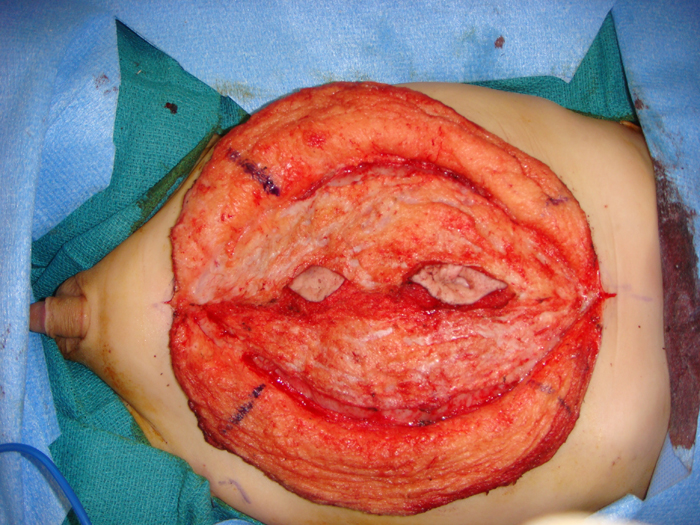
Figura 10: Se ha levantado el colgajo de piel preservando la vascularización de las islas de la piel para el neo ombligo.
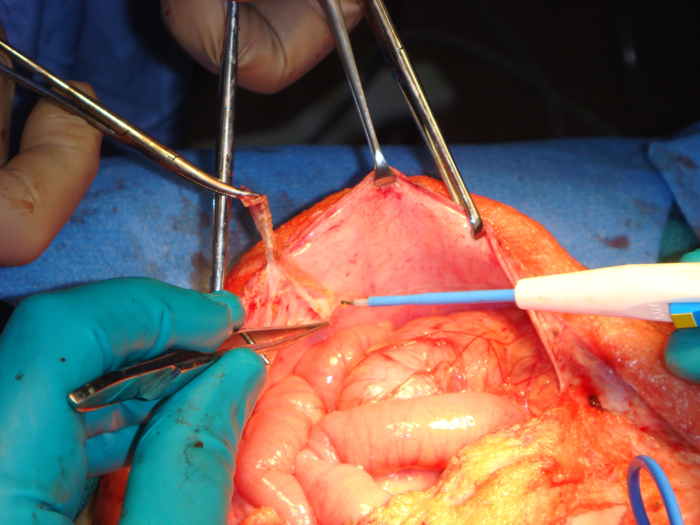
Figura 11: Abdominoplastía, colgajos en isla centrales
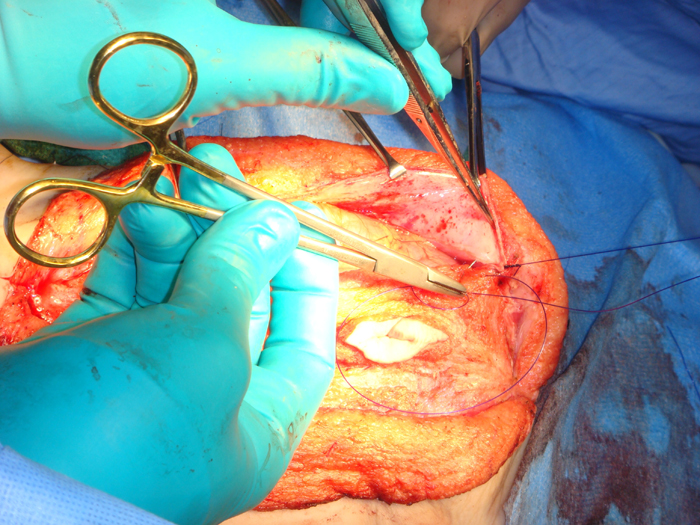
Figura 12: Fijación del colgajo muscular de la isla central al peritoneo parietal posterior
- Platt, W.: Rare Cause of Deficiency of the Abdominal Muscles. Philadelphia Medical Journal, 1: 738, 1898
- Parker, R.: Absence of abdominal muscles in an infant. Lancet, 1: 1252, 1895
- Osler, W.: Congenital absence of the abdominal muscles, with distended and hypertrophied urinary bladder. Bull Johns Hopkins Hospital, 12: 331, 1901
- Nunn, I. N., Stephens, F. D.: The triad syndrome: a composite anomaly of the abdominal wall, urinary system and testes. J Urol, 86: 782, 1961
- Ives, E. J.: The abdominal muscle deficiency triad syndrome–experience with ten cases. Birth Defects Orig Artic Ser, 10: 127, 1974
- Eagle, J. F., Jr., Barrett, G. S.: Congenital deficiency of abdominal musculature with associated genitourinary abnormalities: A syndrome. Report of 9 cases. Pediatrics, 6: 721, 1950
- Frydman, M., Magenis, R. E., Mohandas, T. K. et al.: Chromosome Abnormalities in infants with prune belly anomaly: association with trisomy 18. Am J Med Genet, 15: 145, 1983
- Druschel, C. M.: A descriptive study of prune belly in New York State, 1983 to 1989. Arch Pediatr Adolesc Med, 149: 70, 1995
- Wheatley, J. M., Stephens, F. D., Hutson, J. M.: Prune-belly syndrome: ongoing controversies regarding pathogenesis and management. Semin Pediatr Surg, 5: 95, 1996
- Routh, J. C., Huang, L., Retik, A. B. et al.: Contemporary epidemiology and characterization of newborn males with prune belly syndrome. Urology, 76: 44, 2010
- Reinberg, Y., Shapiro, E., Manivel, J. C. et al.: Prune belly syndrome in females: a triad of abdominal musculature deficiency and anomalies of the urinary and genital systems. J Pediatr, 118: 395, 1991
- Ramasamy, R., Haviland, M., Woodard, J. R. et al.: Patterns of inheritance in familial prune belly syndrome. Urology, 65: 1227, 2005
- Adeyokunnu, A. A., Familusi, J. B.: Prune belly syndrome in two siblings and a first cousin. Possible genetic implications. Am J Dis Child, 136: 23, 1982
- Grenet, P., Le Calve, G., Badoual, J. et al.: [Congenital aplasia of the abdominal wall. A familial case]. Ann Pediatr (Paris), 19: 523, 1972
- Harley, L. M., Chen, Y., Rattner, W. H.: Prune belly syndrome. J Urol, 108: 174, 1972
- Afifi, A. K., Rebeiz, J., Mire, J. et al.: The myopathology of the Prune belly syndrome. J Neurol Sci, 15: 153, 1972
- Garlinger, P., Ott, J.: Prune belly syndrome. Possible genetic implications. Birth Defects Orig Artic Ser, 10: 173, 1974
- Riccardi, V. M., Grum, C. M.: The prune belly anomaly: heterogeneity and superficial X-linkage mimicry. J Med Genet, 14: 266, 1977
- Lockhart, J. L., Reeve, H. R., Bredael, J. J. et al.: Siblings with prune belly syndrome and associated pulmonic stenosis, mental retardation, and deafness. Urology, 14: 140, 1979
- Gaboardi, F., Sterpa, A., Thiebat, E. et al.: Prune-belly syndrome: report of three siblings. Helv Paediatr Acta, 37: 283, 1982
- Feige, A., Fiedler, K., Rempen, A. et al.: [Prenatal diagnosis of prune belly syndrome occurring in siblings in 2 consecutive pregnancies]. Z Geburtshilfe Perinatol, 188: 239, 1984
- Balaji, K. C., Patil, A., Townes, P. L. et al.: Concordant prune belly syndrome in monozygotic twins. Urology, 55: 949, 2000
- Chan, Y. C., Bird, L. M.: Vertically transmitted hypoplasia of the abdominal wall musculature. Clin Dysmorphol, 13: 7, 2004
- Frydman, M., Cohen, H. A., Ashkenazi, A. et al.: Familial segregation of cervical ribs, Sprengel anomaly, preaxial polydactyly, anal atresia, and urethral obstruction: a new syndrome? Am J Med Genet, 45: 717, 1993
- Amacker, E. A., Grass, F. S., Hickey, D. E. et al.: An association of prune belly anomaly with trisomy 21. Am J Med Genet, 23: 919, 1986
- Curry, C. J., Jensen, K., Holland, J. et al.: The Potter sequence: a clinical analysis of 80 cases. Am J Med Genet, 19: 679, 1984
- Hoagland, M. H., Frank, K. A., Hutchins, G. M.: Prune-belly syndrome with prostatic hypoplasia, bladder wall rupture, and massive ascites in a fetus with trisomy 18. Arch Pathol Lab Med, 112: 1126, 1988
- Salihu, H. M., Boos, R., Tchuinguem, G. et al.: Prenatal diagnosis of translocation and a single pericentric inversion 9: the value of fetal ultrasound. J Obstet Gynaecol, 21: 474, 2001
- Fahmy, J., Kaminsky, C. K., Parisi, M. T.: Perlman syndrome: a case report emphasizing its similarity to and distinction from Beckwith-Wiedemann and prune-belly syndromes. Pediatr Radiol, 28: 179, 1998
- Shah, D., Sharma, S., Faridi, M. M. et al.: VACTERL association with Prune-Belly syndrome. Indian Pediatr, 41: 845, 2004
- Levin, T. L., Soghier, L., Blitman, N. M. et al.: Megacystis-microcolon-intestinal hypoperistalsis and prune belly: overlapping syndromes. Pediatr Radiol, 34: 995, 2004
- Weber, S., Mir, S., Schlingmann, K. P. et al.: Gene locus ambiguity in posterior urethral valves/prune-belly syndrome. Pediatr Nephrol, 20: 1036, 2005
- Sutherland, R. S., Mevorach, R. A., Kogan, B. A.: The prune-belly syndrome: current insights. Pediatr Nephrol, 9: 770, 1995
- Pagon, R. A., Smith, D. W., Shepard, T. H.: Urethral obstruction malformation complex: a cause of abdominal muscle deficiency and the "prune belly". J Pediatr, 94: 900, 1979
- Gonzalez, R., Reinberg, Y., Burke, B. et al.: Early bladder outlet obstruction in fetal lambs induces renal dysplasia and the prune-belly syndrome. J Pediatr Surg, 25: 342, 1990
- Lattimer, J. K.: Congenital deficiency of the abdominal musculature and associated genitourinary anomalies: a report of 22 cases. J Urol, 79: 343, 1958
- Rogers, L. W., Ostrow, P. T.: The prune belly syndrome. Report of 20 cases and description of a lethal variant. J Pediatr, 83: 786, 1973
- Moerman, P., Fryns, J. P., Goddeeris, P. et al.: Pathogenesis of the prune-belly syndrome: a functional urethral obstruction caused by prostatic hypoplasia. Pediatrics, 73: 470, 1984
- Rich, D.: The Prune Belly Syndrome. AUA Update Series, 11: 721, 1992
- Mininberg, D. T., Montoya, F., Okada, K. et al.: Subcellular muscle studies in the prune belly syndrome. J Urol, 109: 524, 1973
- O'Kell, R. T.: Embryonic abdominal musculature associated with anomalies of the genitourinary and gastrointestinal systems. Am J Obstet Gynecol, 105: 1283, 1969
- Stephens, F. D., Gupta, D.: Pathogenesis of the prune belly syndrome. J Urol, 152: 2328, 1994
- Wigger, H. J., Blanc, W. A.: The prune belly syndrome. Pathol Annu, 12 Pt 1: 17, 1977
- Deklerk, D. P., Scott, W. W.: Prostatic maldevelopment in the prune belly syndrome: a defect in prostatic stromal-epithelial interaction. J Urol, 120: 341, 1978
- Elder, J. S., Isaacs, J. T., Walsh, P. C.: Androgenic sensitivity of the gubernaculum testis: evidence for hormonal/mechanical interactions in testicular descent. J Urol, 127: 170, 1982
- Fallon, B., Welton, M., Hawtrey, C.: Congenital anomalies associated with cryptorchidism. J Urol, 127: 91, 1982
- Reinberg, Y., Manivel, J. C., Pettinato, G. et al.: Development of renal failure in children with the prune belly syndrome. J Urol, 145: 1017, 1991
- Burke, E. C., Shin, M. H., Kelalis, P. P.: Prune-belly syndrome. Clinical findings and survival. Am J Dis Child, 117: 668, 1969
- Christopher, C. R., Spinelli, A., Severt, D.: Ultrasonic diagnosis of prune-belly syndrome. Obstet Gynecol, 59: 391, 1982
- Hudson, R., Skoog, S.: Prune Belly Syndrome, 5 ed, 2007
- Jennings, R. W.: Prune belly syndrome. Semin Pediatr Surg, 9: 115, 2000
- Woodard, J. R.: The prune belly syndrome. Urol Clin North Am, 5: 75, 1978
- Caldamone, A., Woodard, J. R.: Prune Belly Syndrome, 2009
- Manivel, J. C., Pettinato, G., Reinberg, Y. et al.: Prune belly syndrome: clinicopathologic study of 29 cases. Pediatr Pathol, 9: 691, 1989
- Gearhart, J. P., Lee, B. R., Partin, A. W. et al.: A quantitative histological evaluation of the dilated ureter of childhood. II: Ectopia, posterior urethral valves and the prune belly syndrome. J Urol, 153: 172, 1995
- Palmer, J. M., Tesluk, H.: Ureteral pathology in the prune belly syndrome. J Urol, 111: 701, 1974
- Workman, S. J., Kogan, B. A.: Fetal bladder histology in posterior urethral valves and the prune belly syndrome. J Urol, 144: 337, 1990
- Snyder, H. M., Harrison, N. W., Whitfield, H. N. et al.: Urodynamics in the prune belly syndrome. Br J Urol, 48: 663, 1976
- Kinahan, T. J., Churchill, B. M., McLorie, G. A. et al.: The efficiency of bladder emptying in the prune belly syndrome. J Urol, 148: 600, 1992
- Williams, D. I., Burkholder, G. V.: The prune belly syndrome. J Urol, 98: 244, 1967
- Passerini-Glazel, G., Araguna, F., Chiozza, L. et al.: The P.A.D.U.A. (progressive augmentation by dilating the urethra anterior) procedure for the treatment of severe urethral hypoplasia. J Urol, 140: 1247, 1988
- Shrom, S. H., Cromie, W. J., Duckett, J. W., Jr.: Megalourethra. Urology, 17: 152, 1981
- Kroovand, R. L., Al-Ansari, R. M., Perlmutter, A. D.: Urethral and genital malformations in prune belly syndrome. J Urol, 127: 94, 1982
- Cabral, B., Majidi, A., Gonzalez, R.: Ectopic vasa deferentia in an infant with the prune belly syndrome. J Urol (Paris), 94: 223, 1988
- Orvis, B. R., Bottles, K., Kogan, B. A.: Testicular histology in fetuses with the prune belly syndrome and posterior urethral valves. J Urol, 139: 335, 1988
- Massad, C. A., Cohen, M. B., Kogan, B. A. et al.: Morphology and histochemistry of infant testes in the prune belly syndrome. J Urol, 146: 1598, 1991
- Batata, M. A., Chu, F. C., Hilaris, B. S. et al.: Testicular cancer in cryptorchids. Cancer, 49: 1023, 1982
- Sayre, R., Stephens, R., Chonko, A. M.: Prune belly syndrome and retroperitoneal germ cell tumor. Am J Med, 81: 895, 1986
- Woodhouse, C. R., Ransley, P. G.: Teratoma of the testis in the prune belly syndrome. Br J Urol, 55: 580, 1983
- Woodhouse, C. R., Snyder, H. M., 3rd: Testicular and sexual function in adults with prune belly syndrome. J Urol, 133: 607, 1985
- Burbige, K. A., Amodio, J., Berdon, W. E. et al.: Prune belly syndrome: 35 years of experience. J Urol, 137: 86, 1987
- Kolettis, P. N., Ross, J. H., Kay, R. et al.: Sperm retrieval and intracytoplasmic sperm injection in patients with prune-belly syndrome. Fertil Steril, 72: 948, 1999
- Holder, J. P.: Pathophysiologic and anesthetic correlations of the prune-belly syndrome. AANA J, 57: 137, 1989
- Straub, E., Spranger, J.: Etiology and pathogenesis of the prune belly syndrome. Kidney Int, 20: 695, 1981
- Brinker, M. R., Palutsis, R. S., Sarwark, J. F.: The orthopaedic manifestations of prune-belly (Eagle-Barrett) syndrome. J Bone Joint Surg Am, 77: 251, 1995
- Carey, J. C., Eggert, L., Curry, C. J.: Lower limb deficiency and the urethral obstruction sequence. Birth Defects Orig Artic Ser, 18: 19, 1982
- Joller, R., Scheier, H.: Complete thoracic segmental insensibility accompanying prune belly syndrome with scoliosis. Spine (Phila Pa 1976), 11: 496, 1986
- Geary, D. F., MacLusky, I. B., Churchill, B. M. et al.: A broader spectrum of abnormalities in the prune belly syndrome. J Urol, 135: 324, 1986
- Wright, J. R., Jr., Barth, R. F., Neff, J. C. et al.: Gastrointestinal malformations associated with prune belly syndrome: three cases and a review of the literature. Pediatr Pathol, 5: 421, 1986
- Teramoto, R., Opas, L. M., Andrassy, R.: Splenic torsion with prune belly syndrome. J Pediatr, 98: 91, 1981
- Short, K. L., Groff, D. B., Cook, L.: The concomitant presence of gastroschisis and prune belly syndrome in a twin. J Pediatr Surg, 20: 186, 1985
- Walker, J., Prokurat, A. I., Irving, I. M.: Prune belly syndrome associated with exomphalos and anorectal agenesis. J Pediatr Surg, 22: 215, 1987
- Salihu, H. M., Tchuinguem, G., Aliyu, M. H. et al.: Prune belly syndrome and associated malformations. A 13-year experience from a developing country. West Indian Med J, 52: 281, 2003
- Woodard, J. R., Parrott, T. S.: Reconstruction of the urinary tract in prune belly uropathy. J Urol, 119: 824, 1978
- Ewig, J. M., Griscom, N. T., Wohl, M. E.: The effect of the absence of abdominal muscles on pulmonary function and exercise. Am J Respir Crit Care Med, 153: 1314, 1996
- Alford, B. A., Peoples, W. M., Resnick, J. S. et al.: Pulmonary complications associated with the prune-belly syndrome. Radiology, 129: 401, 1978
- Bellah, R. D., States, L. J., Duckett, J. W.: Pseudoprune-Belly syndrome: imaging findings and clinical outcome. AJR Am J Roentgenol, 167: 1389, 1996
- Yamamoto, H., Nishikawa, S., Hayashi, T. et al.: Antenatal diagnosis of prune belly syndrome at 11 weeks of gestation. J Obstet Gynaecol Res, 27: 37, 2001
- Ellison, L., Cendron, M., Ornvold, K. et al.: Early diagnosis of fetal bladder outlet obstruction. J Pediatr Surg, 35: 513, 2000
- Elder, J.: Intrauterine intervention for obstructive uropathy. Kidney 22: 19, 1990
- Gadziala, N. A., Kawada, C. Y., Doherty, F. J. et al.: Intrauterine decompression of megalocystis during the second trimester of pregnancy. Am J Obstet Gynecol, 144: 355, 1982
- Glazer, G. M., Filly, R. A., Callen, P. W.: The varied sonographic appearance of the urinary tract in the fetus and newborn with urethral obstruction. Radiology, 144: 563, 1982
- Nakayama, D. K., Harrison, M. R., Gross, B. H. et al.: Management of the fetus with an abdominal wall defect. J Pediatr Surg, 19: 408, 1984
- Leeners, B., Sauer, I., Schefels, J. et al.: Prune-belly syndrome: therapeutic options including in utero placement of a vesicoamniotic shunt. J Clin Ultrasound, 28: 500, 2000
- Pescia, G., Cruz, J. M., Weihs, D.: Prenatal diagnosis of prune belly syndrome by means of raised maternal AFP levels. J Genet Hum, 30: 271, 1982
- McLorie, G., Farhat, W., Khoury, A. et al.: Outcome analysis of vesicoamniotic shunting in a comprehensive population. J Urol, 166: 1036, 2001
- Freedman, A. L., Johnson, M. P., Smith, C. A. et al.: Long-term outcome in children after antenatal intervention for obstructive uropathies. Lancet, 354: 374, 1999
- Kramer, S. A.: Current status of fetal intervention for congenital hydronephrosis. J Urol, 130: 641, 1983
- Makino, Y., Kobayashi, H., Kyono, K. et al.: Clinical results of fetal obstructive uropathy treated by vesicoamniotic shunting. Urology, 55: 118, 2000
- Irwin, B. H., Vane, D. W.: Complications of intrauterine intervention for treatment of fetal obstructive uropathy. Urology, 55: 774, 2000
- Soylu, H., Kutlu, N. O., Sonmezgoz, E. et al.: Prune-belly syndrome and pulmonary hypoplasia: a potential cause of death. Pediatr Int, 43: 172, 2001
- Noh, P. H., Cooper, C. S., Winkler, A. C. et al.: Prognostic factors for long-term renal function in boys with the prune-belly syndrome. J Urol, 162: 1399, 1999
- Perlman, M., Levin, M.: Fetal pulmonary hypoplasia, anuria, and oligohydramnios: clinicopathologic observations and review of the literature. Am J Obstet Gynecol, 118: 1119, 1974
- Woodard, J. R.: Lessons learned in 3 decades of managing the prune-belly syndrome. J Urol, 159: 1680, 1998
- Berdon, W. E., Baker, D. H., Wigger, H. J. et al.: The radiologic and pathologic spectrum of the prune belly syndrome. The importance of urethral obstruction in prognosis. Radiol Clin North Am, 15: 83, 1977
- Fallat, M. E., Skoog, S. J., Belman, A. B. et al.: The prune belly syndrome: a comprehensive approach to management. J Urol, 142: 802, 1989
- Woodard, J. R.: Prune belly syndrome. In: Clinical Pediatric Urology. Edited by K. L. R. Kelalis P.P., Belman A.B. Philadelphia: WB Saunders, pp. 805-824, 1985
- Waldbaum, R. S., Marshall, V. F.: The prune belly syndrome: a diagnostic therapeutic plan. J Urol, 103: 668, 1970
- Woodhouse, C. R., Kellett, M. J., Williams, D. I.: Minimal surgical interference in the prune belly syndrome. Br J Urol, 51: 475, 1979
- Randolph, J., Cavett, C., Eng, G.: Surgical correction and rehabilitation for children with "Prune-belly" syndrome. Ann Surg, 193: 757, 1981
- Woodhouse, C. R., Ransley, P. G., Innes-Williams, D.: Prune belly syndrome–report of 47 cases. Arch Dis Child, 57: 856, 1982
- Denes, F. T., Arap, M. A., Giron, A. M. et al.: Comprehensive surgical treatment of prune belly syndrome: 17 years' experience with 32 patients. Urology, 64: 789, 2004
- Tank, E. S., McCoy, G.: Limited surgical intervention in the prune belly syndrome. J Pediatr Surg, 18: 688, 1983
- Randolph, J. G.: Total surgical reconstruction for patients with abdominal muscular deficiency ("prune-belly") syndrome. J Pediatr Surg, 12: 1033, 1977
- Woodard, J. R.: Prune-belly syndrome: a personal learning experience. BJU Int, 92 Suppl 1: 10, 2003
- Perlmutter, A. D.: Reduction cystoplasty in prune belly syndrome. J Urol, 116: 356, 1976
- Bukowski, T. P., Perlmutter, A. D.: Reduction cystoplasty in the prune belly syndrome: a long-term followup. J Urol, 152: 2113, 1994
- Gonzalez, R., De Filippo, R., Jednak, R. et al.: Urethral atresia: long-term outcome in 6 children who survived the neonatal period. J Urol, 165: 2241, 2001
- Reinberg, Y., Chelimsky, G., Gonzalez, R.: Urethral atresia and the prune belly syndrome. Report of 6 cases. Br J Urol, 72: 112, 1993
- Jones, E. A., Freedman, A. L., Ehrlich, R. M.: Megalourethra and urethral diverticula. Urol Clin North Am, 29: 341, 2002
- Heaton, B. W., Snow, B. W., Cartwright, P. C.: Repair of urethral diverticulum by plication. Urology, 44: 749, 1994
- Locke, J. R., Noe, H. N.: Megalourethra: surgical technique for correction of an unusual variant. J Urol, 138: 110, 1987
- Stephens, F. D.: Congenital malformations of the urinary tract. New York: Praeger, pp. xiv, 577 p., 1983
- Smith, E. A., Woodard, J. R.: Prune Belly Syndrome. In: Campbell's Urology, 8 ed. Edited by P. C. Walsh, A. J. Wein, E. D. Vaughan et al. Philadelphia: Saunders, vol. 2, 2002
- Woodard, J. R., Parrott, T. S.: Orchiopexy in the prune belly syndrome. Br J Urol, 50: 348, 1978
- Docimo, S. G., Moore, R. G., Kavoussi, L. R.: Laparoscopic orchidopexy in the prune belly syndrome: a case report and review of the literature. Urology, 45: 679, 1995
- Patil, K. K., Duffy, P. G., Woodhouse, C. R. et al.: Long-term outcome of Fowler-Stephens orchiopexy in boys with prune-belly syndrome. J Urol, 171: 1666, 2004
- Boddy, S. A., Gordon, A. C., Thomas, D. F. et al.: Experience with the Fowler Stephens and microvascular procedures in the management of intraabdominal testes. Br J Urol, 68: 199, 1991
- Smith, C. A., Smith, E. A., Parrott, T. S. et al.: Voiding function in patients with the prune-belly syndrome after Monfort abdominoplasty. J Urol, 159: 1675, 1998
- Randolph, J., Cavett, C., Eng, G.: Abdominal wall reconstruction in the prune belly syndrome. J Pediatr Surg, 16: 960, 1981
- Ehrlich, R. M., Lesavoy, M. A., Fine, R. N.: Total abdominal wall reconstruction in the prune belly syndrome. J Urol, 136: 282, 1986
- Ehrlich, R. M., Lesavoy, M. A.: Umbilicus preservation with total abdominal wall reconstruction in prune-belly syndrome. Urology, 41: 231, 1993
- Monfort, G., Guys, J. M., Bocciardi, A. et al.: A novel technique for reconstruction of the abdominal wall in the prune belly syndrome. J Urol, 146: 639, 1991
- Parrott, T. S., Woodard, J. R.: The Monfort operation for abdominal wall reconstruction in the prune belly syndrome. J Urol, 148: 688, 1992
- Bukowski, T. P., Smith, C. A.: Monfort abdominoplasty with neoumbilical modification. J Urol, 164: 1711, 2000
- Furness, P. D., 3rd, Cheng, E. Y., Franco, I. et al.: The prune-belly syndrome: a new and simplified technique of abdominal wall reconstruction. J Urol, 160: 1195, 1998
- Levine, E., Taub, P. J., Franco, I.: Laparoscopic-assisted abdominal wall reconstruction in prune-belly syndrome. Ann Plast Surg, 58: 162, 2007
- Reinberg, Y., Manivel, J. C., Fryd, D. et al.: The outcome of renal transplantation in children with the prune belly syndrome. J Urol, 142: 1541, 1989
- Kamel, M. H., Thomas, A. A., Al-Mufarrej, F. M. et al.: Deceased-donor kidney transplantation in prune belly syndrome. Urology, 69: 666, 2007
- Fusaro, F., Zanon, G. F., Ferreli, A. M. et al.: Renal transplantation in prune-belly syndrome. Transpl Int, 17: 549, 2004
- Marvin, R. G., Halff, G. A., Elshihabi, I.: Renal allograft torsion associated with prune-belly syndrome. Pediatr Nephrol, 9: 81, 1995