Multicystic Dysplastic Kidney (MCDK) / Multicystic Kidney (MCK)
Abraham Cherian
Great Ormond Street Hospital for Children NHS Trust
→ Enlace a la versión en español

→ Enlace a la versión en español

Multicystic dysplastic kidney is the most common congenital cystic anomaly of the kidney, characterized by multiple non-communicating cysts with minimal intervening tissue on ultrasound scanning. By definition, the affected kidney is non-functioning and is usually associated with an atretic ureter.
Unlike other cystic kidney diseases, such as the dominant and recessive forms of polycystic kidney where there is a clear inherited pattern, MCDK occurs sporadically and is far more common than previously thought as these kidneys remain clinically undetected and asymptomatic. A rare familial form has been reported as autosomal dominant with variable penetrance (1).
Embryology
The embryological basis of this condition is not fully elucidated; however, several hypotheses are presented:
1. Abnormalities of reciprocal induction between the ureteric bud and metanephric blastema is the most popular.
2. Obstruction of the urinary tract during nephrogenesis has also been considered as a possible cause for the development of cystic transformation of the kidney.
3. In-utero viral infections and antiepileptic medications have been implicated in some reports (2,3).
Incidence
The incidence is about 1 in 4000 and is more common in males (2.4:1). Females are twice as likely to develop bilateral MCDK. The left side is more common than the right.
Unilateral (UMCDK), bilateral (BMCDK), and segmental (SMCDK) are recognized forms of involvement. Bilateral disease is incompatible with life.
Pathology
Macroscopy: On gross appearance, 2 types are described: solid cystic dysplasia and the hydronephrotic forms. The former has smaller and fewer cysts with more stroma and the latter has cysts of varying sizes and a recognizable but dilated renal pelvis with very little intervening stroma. The vascular pedicle is hypoplastic or absent. The ureter is usually atretic if present. Previously thought to be non-communicating, these cysts have, in several studies, proven to have communications with each other both on microscopy and radiological intracystic injections of contrast medium (4-6)
Microscopy: There are components of both abnormal (dysplastic) and normal nephrogenesis. Dysplastic elements include primitive ducts with concentric rings of collagen, and occasional smooth muscle cells surrounding it. Others are metaplastic cartilage, primitive ducts and glomeruli, intervening mesenchyme and fibrous tissue. Cysts are lined by flattened squamous or columnar epithelium that have originated from all segments of the nephron, from glomerulus to the collecting duct.
Clinical Features
Historically, a palpable mass per abdomen was the commonest presentation. This has been surpassed by increasing detection on antenatal ultrasound scanning and forms the largest group of patients currently. There is some evidence to suggest that unilateral renal agenesis may represent an MCDK that has regressed (7). Pain or discomfort in the flank, haematuria, UTI, and hypertension are other presentations postnatally.
Segmental MCDK is a rare form and can occur in horseshoe kidneys and moieties of duplex systems. A large segmental MCDK may obscure a much smaller functioning segment where radioactive isotope study is the only method of identification.
Other ipsilateral associations that have been reported are vesico-ureteric reflux (VUR), vesico-uereteric junction obstruction (VUJO) and ureterocoeles. In a duplex system, a pelvi-ureteric junction obstruction (PUJO) in the other moiety has also been noted.
On the contralateral side, VUR is the most common anomaly, occurring in up to 25%; however these cases are mostly under grade 3 reflux. Other uncommon findings are PUJO and VUJO.
Investigations
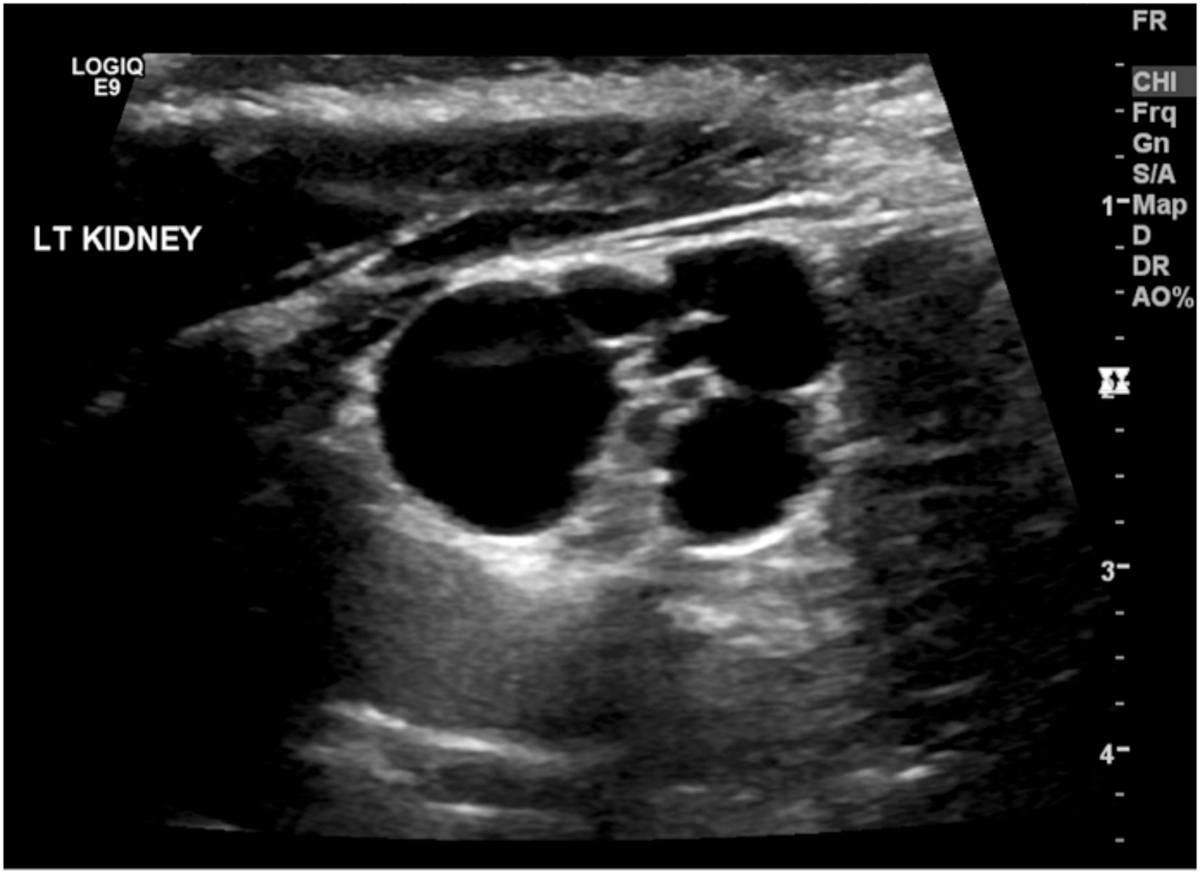
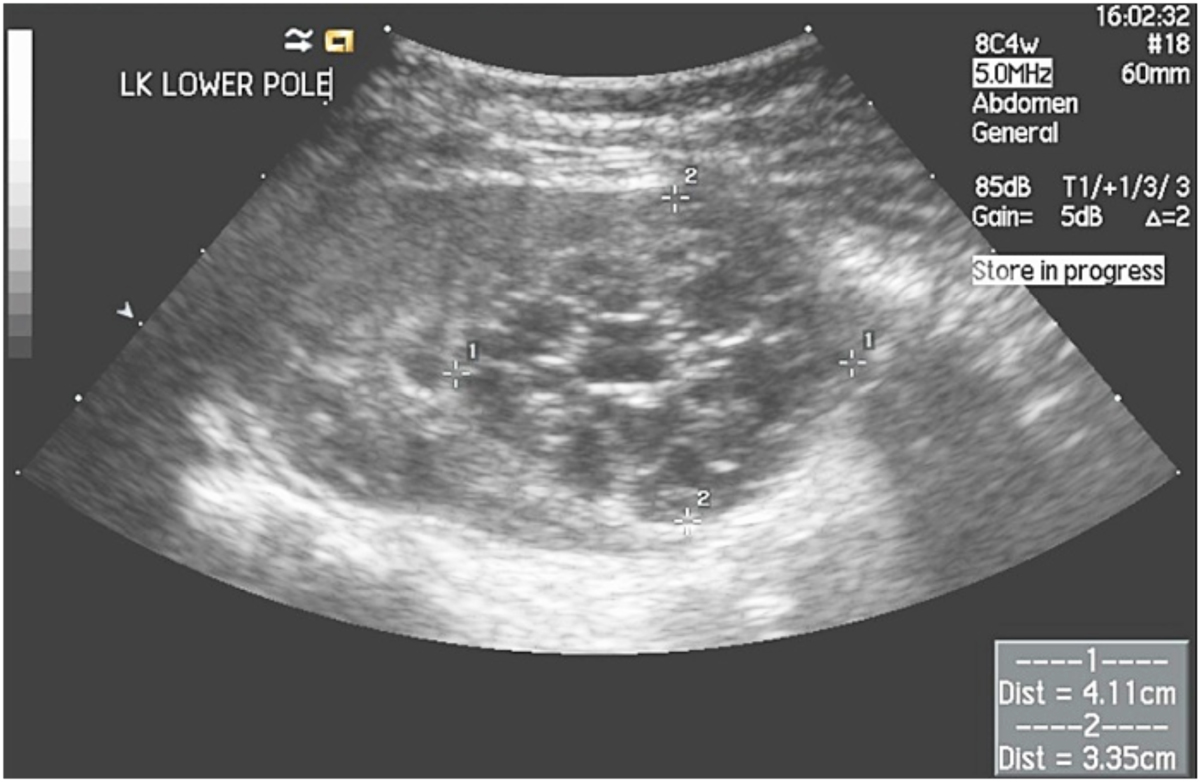
Diagnosis is considered by ultrasound findings demonstrating non-communicating cysts (Figure 1.1) with very little intervening parenchyma. It may be confirmed by absence of uptake on radio-isotope scanning such as a Tc99m-DMSA or Tc99m-MAG3 scan.
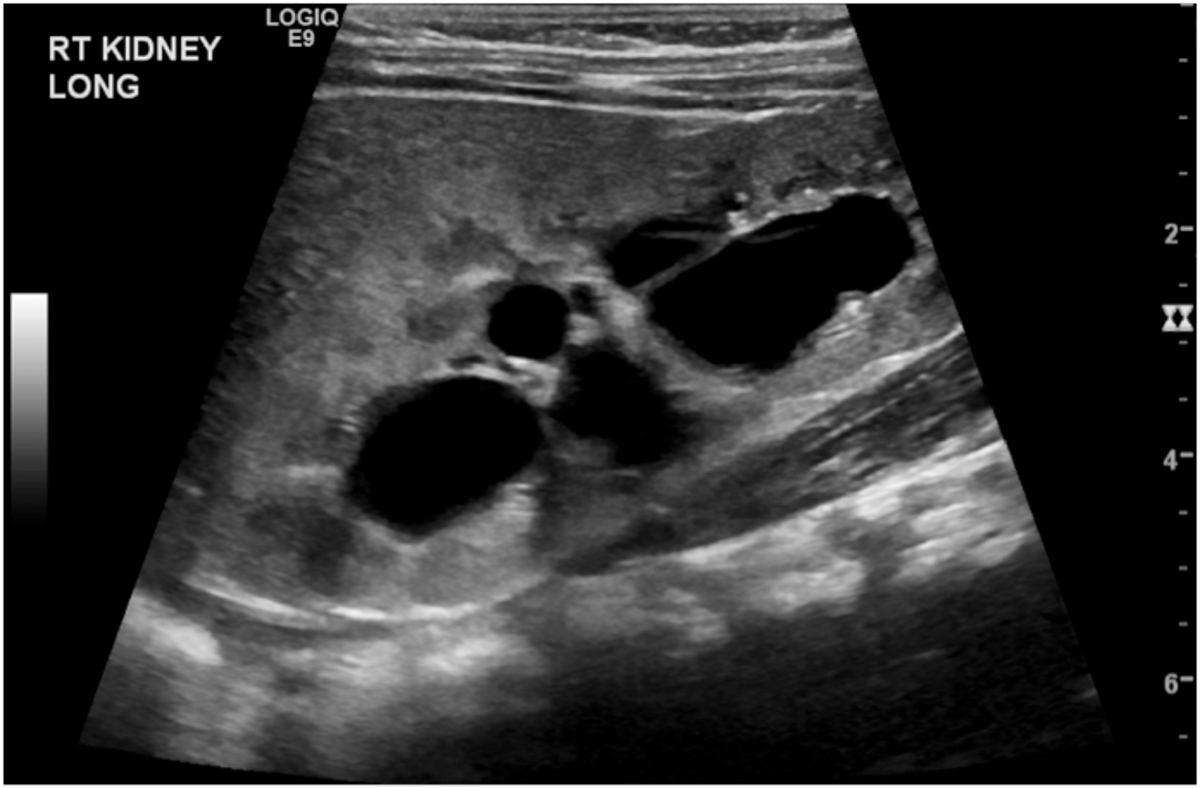
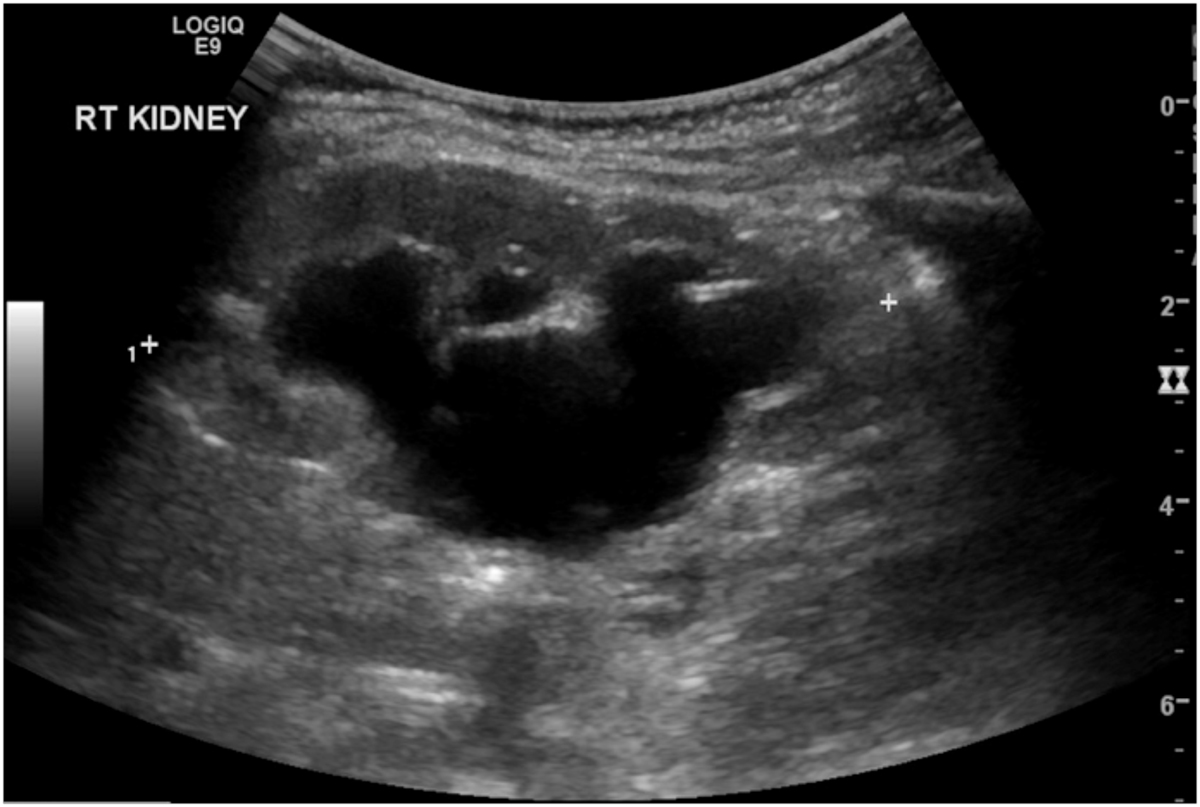
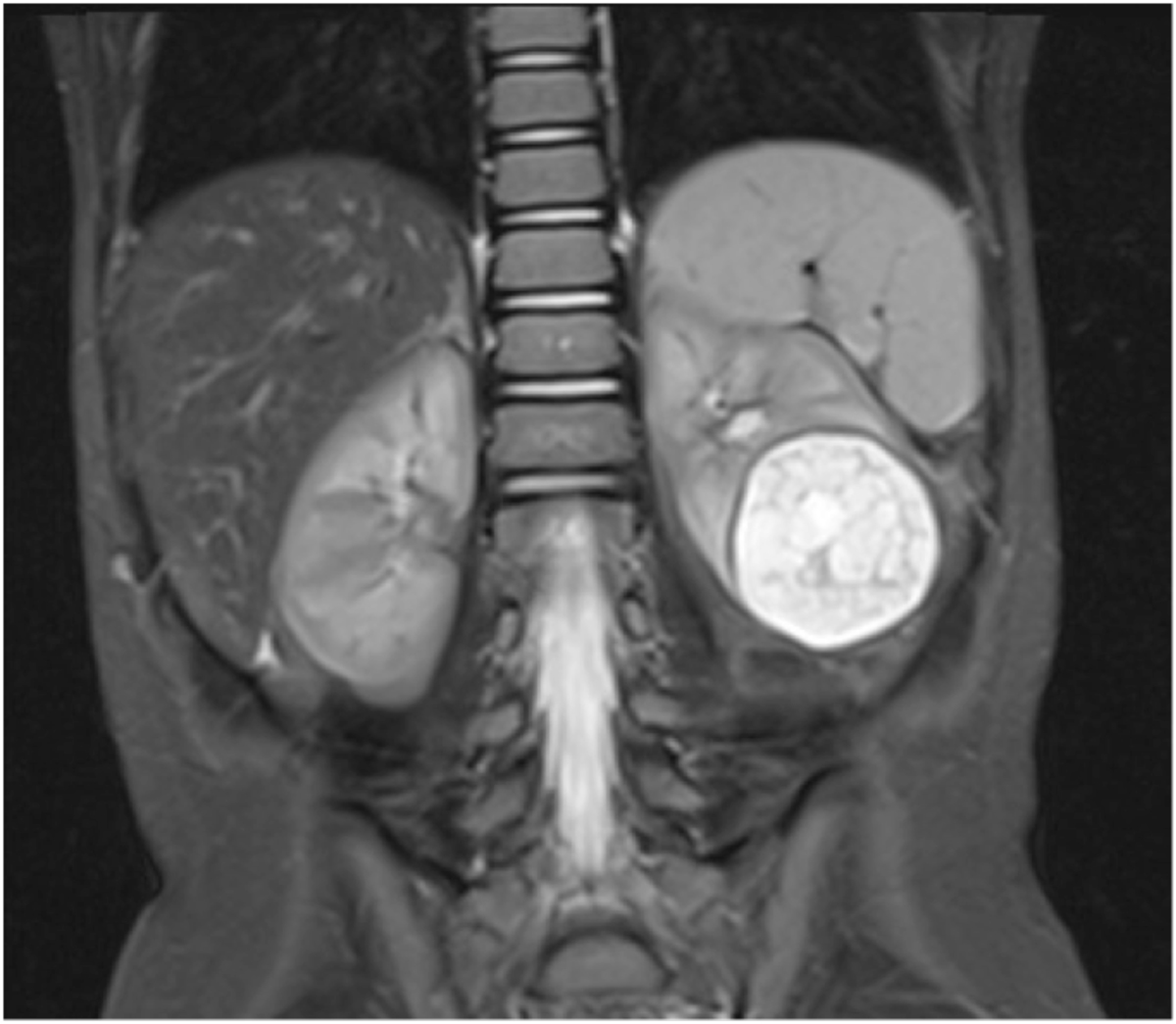
The conditions that mimic MCDK include severe intrarenal hydronephrosis due to a pelvi-ureteric junction obstruction(Figures 1.2, 1.3). The latter condition usually has some intervening and surrounding parenchyma, communicating cysts on ultrasound with some uptake on the radioisotope scan. Others include a cystic nephroma (Figure 2.1), though this is rare and difficult to distinguish from segmental MCDK, and may MRI for identification. (Figure 2.2). In some cases a percutaneous or excisional biopsy may be necessary.
Cystograms are unnecessary in isolated MCDK with a normal contralateral kidney. Obtaining a glomerular filtration rate is recommended as a baseline and to confirm the function of the contralateral kidney.
Management
A uniform approach in the management of MCDK has not been universally established; however various aspects need consideration, namely spontaneous involution, risk of hypertension, vesico-ureteric reflux, risk of cancer and follow-up protocols.
A comprehensive literature review was conducted by Cambio et al in 2008 and forms the basis of discussion to guide management outlined below (8). This review included the analysis of 105 publications and data on another 900 MCDKs from the MCDK Registry (9). Outcomes studied were vesico-ureteric reflux, hypertension, malignancy and need for surgery.
Overall, involution—defined as being undetectable by ultrasound—was noted in 60%; 20% were undetectable by 3 years and 50% by 5 years. Vesico-ureteric reflux occurred in 25%, but this was into the contralateral kidney and more than 90% were grade 3 or less. The risk of urinary tract infection was related to reflux or abnormalities other than the MCDK. Compared to the general population, there was no increased risk of hypertension; however, this may be considered to be an indication for removal. In more than two-thirds, hypertension persists despite surgical removal.
In 1997, Beckwith et al reported the calculated the risk of Wilms tumor in MCDK as less than 1 in 2000 (10). Noe et al came to the same conclusion quite independently with a number needed to treat as 2000 to prevent the occurrence of one Wilms tumor (11). Wacksmann and Phipps in their preliminary report on the MCDK Registry recorded only 2 Wilms tumors spanning a 25-year period (9).
There were 10 reported cases of Wilms tumor in the review and all were before the age of four (8). Seven of the 10 presented with a palpable mass. There has been no reported Wilms in an involuted MCDK. There were 6 reports of renal cell carcinoma (RCC) associated with MCDK between 15 and 44 years of age. However, the accuracy of pathology is debatable as these were not subjected to external pathology review as with Wilms tumors. It is possible that some were cystic RCCs. There were no cases of Wilms tumor from the published data of the MCDK Registry, which included 260 cases managed non-operatively. The same inference was gathered from the prospective data collected by the Trent & Anglia MCDK Study Group (12).
Indications for Surgery
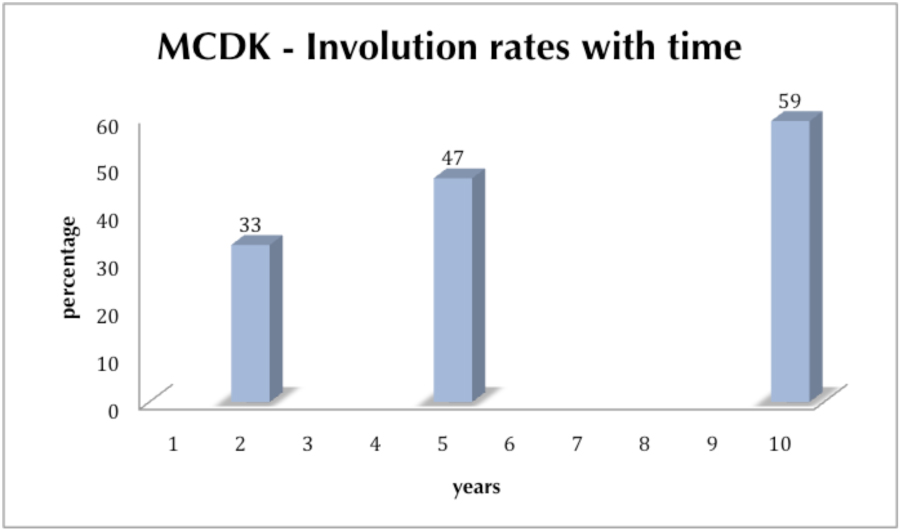
There are no predictive factors to determine spontaneous resolution. There are some anecdotal reports to suggest that a size of greater than 6 centimeters at one year of age is unlikely to resolve spontaneously. Up to 40% of MCDKs were still being followed beyond the age of 10 (Figure 1.0).
A clear discussion with the family should be explored in select cases with the offer of removal as this is curative, with low morbidity and is cost-effective without the anxiety of ongoing surveillance. A progressively decreasing size can be clearly managed non-operatively.
There are no clear guidelines with regard to surveillance; however, it seems unnecessary beyond the age of four for malignancy.
Surgical removal of MCDK can be accomplished by both conventional open and laparoscopic techniques. Large MCDKs may need cysts decompressed to facilitate removal. Both trans-peritoneal and retro-peritoneoscopic approaches are well established techniques. Follow-up after surgery maybe up to a year and include an ultrasound and serum creatinine from which an estimated glomerular filtration rate can be obtained. Routine GFR measurements are probably unnecessary in the absence of any underlying risk factors (13).
Summary points
References
1. Watanabe T, Yamazaki A, Kurabayashi T, Hanaoka J. (2005) Familial multicystic dysplastic kidney Pediatr Nephrol 2005;20:1200.
2. Petrikovsky BM, Lipson SM, Kaplan MH. Viral studies on amniotic fluid from fetuses with and without abnormalities detected by prenatal sonography. J Reprod Med 2003;48:230-232.
3. Carta M, Cimador M, Giuffre M, Sergio M, Di Pace MR, De Grazia E, Corsello G. Unilateral multicystic dysplastic kidney in infants exposed to antiepileptic drugs during pregnancy. Pediatr Nephrol 2007;22:1054-1057.
4. Felson B, Cussen LJ. The hydronephrotic type of congenital multicystic disease of the kidney. Semin Roentgenol 1975;10:113-123.
5. Glassberg KI, Kassner EG. Ex vivo intracystic contrast studies of multicystic dysplastic kidneys. J Urol 1998;160:1204-1206.
6. Dewan PA, Goh DW. A study of the radiological anatomy of the multicystic kidney. Pediatr Surg Int 1994;9:368-372.
7. Hitchcock R, Burge DM. Renal agenesis: an acquired condition?. J Pediatr Surg 1994;29:454-455.
8. Cambio AJ, Evans CP, Kurzrock EA. Non-surgical management of multicystic dysplastic kidney. BJU Int 2008;101:804-808.
9. Wacksman J, Phipps L. Report of the MCDK Registry: preliminary findings J Urol 1993;150:1870-1872.
10. Beckwith JB. Wilms tumor and multicystic dysplastic kidney disease. Editorial Comment. J Urol 1997;158:2259-2260.
11. Noe HN, Marshall JH, Edwards OP. Nodular renal blastema in the multicystic kidney. J Urol 1989;142:486-488.
12. Aslam M, Watson AR, on behalf of the Trent & Anglia MCDK Study Group. (2006). Unilateral multicystic dysplastic kidney: long term outcomes. Arch Dis Child 91:820-823.
13. Godbole PP, Wilcox DT, Mushtaq I. Follow-up after unilateral nephrectomy in children: is an estimate of glomerular filtration rate necessary? BJU Int 2005;95:635-637.
 |
 |
 |
 |
 |
 |
 |
 |