









Enfermedad Renal Quística
Paul JD Winyard y Karen L Price
Unidad de Nefrología-Urología, Instituto UCL de Salud Infantil, Londres, Reino Unido
Autor correspondiente: [email protected]
Traducido y editado desde el original al español | Enlace a la versión en inglés
Dra Cinthia Galvez Alegria, Dr. Miguel Castellan
Miami Children's Hospital, Jackson Memorial Hospital, Miami, Fl, USA
Visión general
Por definición, los riñones quísticos contienen quistes en parte o todo el tejido renal normal, pero existen muchos tipos y una marcada heterogeneidad clínica. Para la clasificación, los riñones quísticos se pueden dividir en causas genéticas o no genéticas, pero las nuevas tecnologías están reduciendo rápidamente la proporción en el último grupo y está claro que muchos casos aparentemente esporádicos tienen mutaciones subyacentes. La enfermedad renal poliquística es la forma genética clásica y se divide en herencia autosómica dominante (ADPKD) y recesiva (ARPKD). Las mutaciones de ADPKD son mucho más comunes (1 en 6-800) que ARPKD (1 en 5.000 frecuencia de mutación, lo que equivale a 1 en 20.000 enfermedades) pero es la última la que tiene más probabilidades de causar problemas con la presentación prenatal y temprana con enfermedad renal crónica durante infancia. Las otras formas principales heredadas son las recientemente denominadas "ciliopatías", donde los defectos en el desarrollo o la función de los cilios primarios conducen a quistes renales, defectos de lateralidad y diversas patologías. Los ejemplos incluyen los síndromes de Bardet-Biedl, la nefrona tisis y hay un movimiento para incluir la PKD en este grupo también. Los riñones quísticos esporádicos caen en el espectro displásico multiquístico / quístico donde los quistes a menudo son parte de un defecto de desarrollo más amplio en los riñones y el tracto urinario. Estas a veces se denominan anomalías congénitas del riñón y del tracto urinario (CAKUT).
Diagnóstico prenatal
Los riñones quísticos se detectan fácilmente con ultrasonido, por lo que cada vez se diagnostican más en la exploración prenatal (Winyard y Chitty 2008). Sin embargo, hay una dificultad en que las primeras etapas de cualquier desarrollo de quistes o los múltiples quistes pequeños de ARPKD están por debajo de la resolución de la ecografía. Por lo tanto, puede haber apariencias no específicas en lugar de quistes francos, que incluyen uno o más de tamaño aumentado, eco textura brillante y pérdida de diferenciación cortico-medular. El ADPKD también puede presentarse así en el período del neonatal, pero de manera más clásica presenta algunos quistes grandes que se multiplican y se expanden con el tiempo. Los riñones displásicos multiquísticos también tienen quistes de moderados a grandes en su presentación, pero hay poco tejido renal normal entre ellos.
ADPKD
ADPKD representa alrededor del 5-10% de los pacientes adultos en diálisis o con un trasplante renal en el mundo desarrollado, pero rara vez causa problemas en la infancia (Fick-Brosnahan et al. 2001). El 90% de los pacientes tiene un padre afectado, y el diagnóstico puede confirmarse virtualmente al encontrar quistes en el mismo. Sin embargo, los quistes renales se desarrollan progresivamente durante muchas décadas, por lo que uno debe tener cuidado con los resultados negativos si los padres son jóvenes. Una guía útil es que alrededor del 95% de las personas afectadas tendrán quistes cuando alcancen los 30 (Liapis y Winyard 2007). Las complicaciones renales incluyen cálculos, infecciones, dolor en el flanco y hematuria macroscópica, pero también existe un riesgo de hipertensión, quistes hepáticos y pancreáticos, y aneurismas cerebrales.
Las mutaciones en PKD1 o PKD2 causan más del 98% de ADPKD (Harris y Rossetti 2010). Un tercer locus (PKD3) ha sido postulado, pero nunca probado. Las mutaciones de la PKD1 tienen un inicio más temprano de los síntomas en comparación con la PKD2, pero ambos genes exhiben una heterogeneidad alélica extensa y manifestaciones clínicas variables, quizás atribuibles a los antecedentes genéticos y las diferencias ambientales que modifican la patogénesis (Fain et al. 2005). PKD1 se encuentra en el cromosoma 16p13.3 y codifica una proteína de 460 kDa, policistina 1. PKD2 se asigna a 4q13-q23 y codifica policistina 2, un canal de calcio que interactúa con su contraparte más grande en un complejo de policistina transmembrana en la membrana plasmática. La localización incluye cilios, pero ambas policistinas también se encuentran en muchos otros sitios subcelulares, incluidas las uniones de células basales y laterales (Wilson 2004)
ARPKD
La ARPKD es mucho menos común que la ADPKD, pero es más probable que se presente antes del nacimiento o en la infancia, por lo que a menudo se la conocía como "PKD infantil" antes de que se definiera su herencia y genética. Este término confuso no se debe usar ya que en la actualidad existe una gran cantidad de la enfermedad dominante diagnosticada en la niñez debido a las pruebas genéticas familiares. Generalmente, la ARPKD se presenta en forma prenatal debido a riñones agrandados y brillantes, y existe una alta probabilidad de muerte perinatal por desarrollo pulmonar inmaduro si hay oligo o anhidramnios severos antes de las 24 semanas de gestación (Winyard y Chitty 2001). Después del nacimiento, los niños afectados pueden ser diagnosticados de manera fortuita debido al agrandamiento de los riñones, pero es más probable que presenten síntomas y signos de insuficiencia renal. Denota, la hipertensión es común en ARPKD y puede requerir múltiples medicamentos para controlarla. La fibrosis hepática también puede convertirse en un problema a medida que el niño crece. Alrededor del 85% de los pacientes con ARPKD sobreviven al período perinatal, y las cifras históricas de los EE.UU. sugieren un 78% al año y un 75% de supervivencia a los 5 años (Guay-Woodford y Desmond 2003). Los estudios paralelos del Reino Unido sugieren una supervivencia renal actuarial del 86% al año y del 67% a los 15 años para aquellos pacientes que sobreviven el primer mes (Roy et al. 1997).
Las mutaciones en el riñón poliquístico y el gen de la enfermedad hepática 1 (PKHD1) causan ARPKD; este se encuentra en el cromosoma 6p21 y codifica una proteína llamada fibrocistina (o poliductina de diferentes autores). Existe una gran cantidad de mutaciones conocidas diseminadas por todo el gen, y la mayoría de los pacientes son heterocigotos compuestos. Existe un alto riesgo de presentación fetal y muerte neonatal si el niño tiene dos mutaciones truncantes (Bergmann et al. 2005). La fibrocistina se localiza fuertemente en el cilio primario de las células epiteliales renales, y existe evidencia de que esto también puede formar parte del complejo policistina más amplio (Wang et al. 2007). Los quistes solo se derivan de los conductos colectores en ARPKD, pero pueden provenir de todos los segmentos de nefrona en ADPKD (¡aunque el 85-90% todavía surge de los conductos colectores!).
Quistes simples
Los quistes renales simples suelen ser solitarios y no tienen consecuencias funcionales. Su etiología es desconocida, pero no se heredan. Los quistes se vuelven más comunes a medida que las personas envejecen; de hecho, se estima que casi un tercio de los mayores de 70 tienen al menos un quiste simple. Los niños rara vez tienen quistes simples, pero hay informes de casos de quistes aislados con función renal normal, ecografía irreprochable del resto del riñón y tracto urinario y no hay evidencia de hipertensión (Murthi et al. 2001). La mayoría de los casos son asintomáticos y solo se detectan por casualidad cuando se realiza una ecografía abdominal / renal para otros fines. Sin embargo, el dolor puede ocurrir de manera secundaria a los quistes y ocasionalmente se infectan o comienzan a sangrar.
Displasia quística y riñones displásicos multiquísticos
La displasia implica desarrollo anormal y / o componentes tisulares, lo que resalta inmediatamente una diferencia con la PKD porque los quistes en esta última se generan a partir de partes normales de la nefrona, mientras que hay reducción a cero de "tejidos normales" dentro de los órganos displásicos. En cambio, los riñones displásicos contienen nefrones diferenciados / ramificados y conductos colectores con aumento del estroma y músculo liso (Winyard y Chitty 2008). Ocasionalmente, también hay áreas metaplásicas de cartílago. Los riñones displásicos son comunes, con mal desarrollo unilateral que ocurre en 1 de cada 1000 y bilateral en 1 de cada 5000, aunque solo un subconjunto de estos contiene quistes significativos. Los riñones displásicos quísticos representan un extremo de este espectro, a menudo contienen áreas con nefronas relativamente normales adyacentes a regiones displásicas con quistes pequeños a moderados. Los riñones displásicos multiquísticos son el otro extremo, y comprenden solo una colección de quistes sin tejido renal funcional. A menudo están conectados a uréteres atrésicos, y muchas de las características moleculares de la displasia pueden replicarse por obstrucción en animales experimentales, pero es probable que las influencias genéticas y los factores ambientales también desempeñen un papel en la patogénesis (Yang et al. 2001).
La ecografía prenatal a menudo visualiza los riñones displásicos en la exploración de rutina de 20 semanas, generalmente como riñones grandes y brillantes, con o sin espacios quísticos. También pueden ser pequeños, pero existe un sesgo de determinación para los más grandes, ya que son más fáciles de visualizar y los riñones más pequeños pueden ser enmascarados (o atribuidos a) dificultades técnicas. Los riñones displásicos multiquísticos tienen aspectos característicos de múltiples quistes no conectados de paredes delgadas en un riñón agrandado con un contorno irregular no reniforme. A veces son difíciles de distinguir de la hidronefrosis grave con dilatación calicial, pero la falta de una pelvis renal normalmente situada y la ausencia de flujo arterial mediante ecografía Doppler color puede ser diagnóstica. Muchos riñones displásicos intervienen a medida que el feto / niño crece (Rickwood et al. 1992; Rottenberg et al. 1997) y pueden desaparecer antes o después del nacimiento. El factor crítico para la función renal a largo plazo es si el riñón contralateral sufre una hipertrofia compensatoria (Rudnik-Schoneborn et al. 1998).
La mayoría de los riñones displásicos parecen esporádicos, aunque la historia familiar de malformaciones renales o urinarias se produce históricamente en el 10-15% de los casos (McPherson et al. 1987). La causa genética más común son las mutaciones del gen TCF2 que codifica la proteína hepatocito nuclear factor (HNF) 1β. Esto se informó inicialmente con los riñones quísticos y la diabetes de inicio en la madurez de los jóvenes, pero el espectro de malformaciones ahora incluye los riñones displásicos quísticos, hipoplásicos, poliquísticos y glomerulocísticos, así como los riñones solitarios en los que puede haber participado (Edghill et al. 2006). En análisis recientes de poblaciones seleccionadas, se informaron mutaciones de TCF2 en el 29% de una serie de 62 pacientes con riñones brillantes diagnosticados con antenas (Decramer et al. 2007) y el 23% en nuestro hospital de referencia de especialistas (Adalat et al. 2009). También se han informado mutaciones con riñones displásicos en muchos otros genes en los últimos años, incluyendo PAX-2 (síndrome de coloboma renal), Uroplakins, BMP4, SIX-1 y 2, EYA1 (síndrome de Branchio-oto-renal) y SALL1 (Síndrome de Townes-Brocks) (Jenkins et al. 2005; Weber et al. 2006; Weber et al. 2008).
También hay varias enfermedades glomerulocísticas hereditarias que combinan elementos variables de displasia (desde hipoplasia leve hasta displasia quística franca) con quistes claros que surgen de la cápsula de Bowman. Algunas veces se pueden confundir con ADPKD, pero muchos de los casos sindrómicos, como el síndrome oro-facial digital, tienen anomalías clásicas en otros órganos que apuntan hacia el verdadero diagnóstico. BMP4, SIX-1 y 2, EYA1 (síndrome de Branchio-oto-renal) y SALL1 (síndrome de Townes-Brocks) (Jenkins et al. 2005; Weber et al. 2006; Weber et al. 2008). También hay varias enfermedades glomerulocísticas hereditarias que combinan elementos variables de displasia (desde hipoplasia leve hasta displasia quística franca) con quistes claros que surgen de la cápsula de Bowman. Algunas veces se pueden confundir con ADPKD, pero muchos de los casos sindrómicos, como el síndrome oro-facial digital, tienen anomalías clásicas en otros órganos que apuntan hacia el verdadero diagnóstico. BMP4, SIX-1 y 2, EYA1 (síndrome de Branchio-oto-renal) y SALL1 (síndrome de Townes-Brocks) (Jenkins et al. 2005; Weber et al. 2006; Weber et al. 2008). También hay varias enfermedades glomerulocísticas hereditarias que combinan elementos variables de displasia (desde hipoplasia leve hasta displasia quística franca) con quistes claros que surgen de la cápsula de Bowman. Algunas veces se pueden confundir con ADPKD, pero muchos de los casos sindrómicos, como el síndrome oro-facial digital, tienen anomalías clásicas en otros órganos que apuntan hacia el verdadero diagnóstico. También hay varias enfermedades glomerulocísticas hereditarias que combinan elementos variables de displasia (desde hipoplasia leve hasta displasia quística franca) con quistes claros que surgen de la cápsula de Bowman. Algunas veces se pueden confundir con ADPKD, pero muchos de los casos sindrómicos, como el síndrome oro-facial digital, tienen anomalías clásicas en otros órganos que apuntan hacia el verdadero diagnóstico. También hay varias enfermedades glomerulocísticas hereditarias que combinan elementos variables de displasia (desde hipoplasia leve hasta displasia quística franca) con quistes claros que surgen de la cápsula de Bowman. Algunas veces se pueden confundir con ADPKD, pero muchos de los casos sindrómicos, como el síndrome oro-facial digital, tienen anomalías clásicas en otros órganos que apuntan hacia el verdadero diagnóstico.
La probabilidad de desarrollar insuficiencia renal crónica en pacientes con displasia bilateral es difícil de predecir antes del nacimiento, pero existe una clara correlación entre la función renal en la primera infancia y el resultado a largo plazo (Ismaili et al. 2001; Van Dyck et al. 1998). Cabe destacar, sin embargo, que la maduración renal postnatal normal puede retrasarse varios años en los riñones displásicos (González et al. 2007). El pronóstico de la displasia asociada con malformaciones del tracto urinario inferior no se puede estimar con precisión hasta que se corrijan las anomalías (Drozdz et al. 1998; Godley et al. 2001).
Los informes anecdóticos sugirieron que los riñones displásicos corren el riesgo de desarrollar hipertensión y tumores, pero las revisiones sistemáticas recientes han sugerido que estas complicaciones son muy poco frecuentes (Narchi 2005a; Narchi 2005b), y no hay una razón basada en la evidencia para la nefrectomía electiva a menos que haya complicaciones como las frecuentes infecciones del tracto urinario.
Enfermedades basadas en la ciliopatía
Hay un número creciente de condiciones que se clasifican como ciliopatías debido a que los genes mutados e involucrados codifican proteínas que se localizan en el cilio primario (Hildebrandt et al. 2009). Estos son orgánulos con forma de dedos cerrados por una membrana que se proyectan desde la célula, generalmente en la superficie apical o hacia una luz, conducto o vía aérea (Ong y Wheatley 2003). Nuevamente, la terminología es confusa porque la mayoría de los libros de texto incluyen una discusión de enfermedades como el síndrome de Kartagener y dicen que esto se debe a la discinesia ciliar primaria, ¡pero el defecto no está en absoluto en los cilios primarios!, el defecto primario se encuentra en los cilios móviles que tienen la disposición clásica '9 + 2' de microtúbulos estructurales necesarios para el batido coordinado para eliminar las secreciones respiratorias. Los verdaderos cilios primarios tienen una disposición '9 + 0' que carece de los microtúbulos centrales y estos tienen múltiples funciones en la mecanosensación, la fotorecepción y la quimiosensación. Es el primero de estos que vincula las ciliopatías con las enfermedades quísticas renales: el flujo de fluido fisiológico a través de la superficie apical de las células renales dobla los cilios e induce la entrada de calcio, y esto se perturba cuando se mutan los genes de la ciliopatía. Los efectos posteriores incluyen cambios en la diferenciación celular y la polaridad, con la perturbación de vías icónicas como la señalización de Hedgehog y Wnt (Winyard y Jenkins 2011). El flujo de fluido fisiológico a través de la superficie apical de las células renales dobla los cilios e induce la entrada de calcio, y esto se ve perturbado cuando los genes de la ciliopatía están mutados.
Los genes PKD se expresan en el cilio primario y claramente tienen una formación masiva de quistes y destrucción de tejidos normales, pero muchas de las otras condiciones en la lista de ciliopatía tienen sus principales anomalías en otros sistemas de órganos como el corazón, los ojos o el sistema nervioso; por lo tanto, no se analizarán más aquí, pero hay varias revisiones recientes excelentes (Waters y Beales 2011). A modo de referencia, la lista actual incluye el síndrome de Bardet-Biedl, la nefrona- tisis, el síndrome de Alstrom, el síndrome orofaciodigital, el síndrome de Senior-Loken y el síndrome de Usher.
Tratamiento de las enfermedades quísticas renales
Actualmente no existen tratamientos específicos dirigidos al desarrollo de los quistes. Por lo tanto, el pilar del tratamiento para todas estas afecciones es la atención de apoyo para regular la presión arterial y manejar las complicaciones de la enfermedad renal crónica (ERC). El bloqueo de la vía renina-angiotensina que usa inhibidores de la ECA y bloqueadores de receptores es una terapia de rutina en la mayoría de la ERC, particularmente cuando existe una proteinuria significativa, y recientemente se ha demostrado que frena el deterioro renal en niños (Wuhl et al. 2009). La cirugía solo es realmente útil para corregir anomalías urológicas, y la eliminación de quistes o la aspiración rara vez se utiliza, ya que los quistes se acumulan rápidamente. La nefrectomía en ocasiones se justifica antes del trasplante si los riñones son masivos, pero la mayoría de los órganos tienden a ser fibrosos y se vuelven más pequeños a medida que se alcanza la etapa final.
El trabajo experimental con animales en la PKD ha descubierto una serie de nuevos tratamientos potenciales para algunas afecciones en los últimos 5 años. Estos incluyen la modulación del sistema de vasopresina usando tolavaptán / agentes similares o una mayor ingesta de agua; bloqueo de las vías de mTOR que usan drogas similares a la rapamicina y la somatostatina. La lista actualizada de ensayos se puede ver en http://www.pkdcure.org/Research/ClinicalTrials/ActiveRecruiting.aspx Se espera que uno o más de estos puedan ser exitosos y que estos tratamientos puedan ser probados para otros quistes pediátricos y enfermedades en el futuro.
Figura
Hematoxilina y secciones teñidas con eosina de riñones quísticos pediátricos. Las vistas de la corteza se muestran en la columna izquierda y la médula en la derecha. A y B) PKD autosómica dominante con algunos quistes en la corteza y la médula que surgen de todos los segmentos de nefrona. Esto fue desde una etapa temprana, de ahí la arquitectura bien conservada de los glomérulos, túbulos y conductos colectores que rodean normalmente. C y D) PKD autosómica recesiva con numerosos quistes alargados que atraviesan la corteza y la médula; tenga en cuenta que todos los glomérulos parecen tener un espacio de Bowman normal, no dilatado, ya que los quistes solo surgen de los conductos colectores en esta condición. E y F) Enfermedades renales glomerulocísticas con displasia marcada; note la interrupción significativa de la diferenciación renal y los mechones glomerulares visibles en prácticamente todos los quistes.
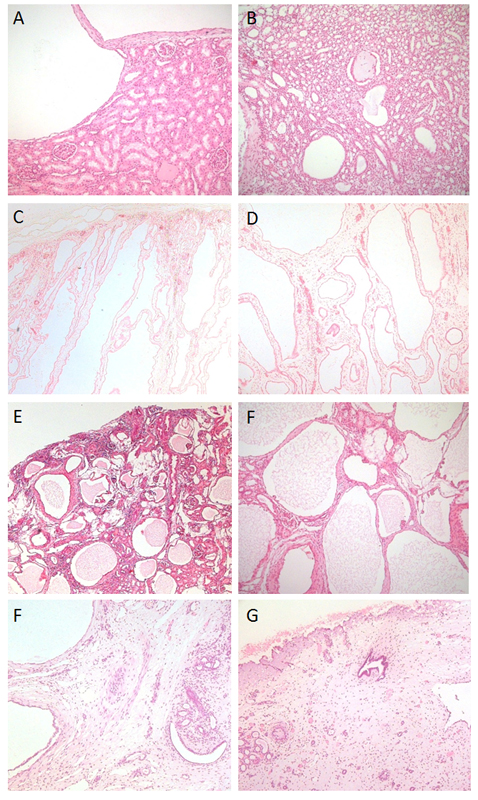
Figura 1
Referencias
Adalat, S., Woolf, A.S., Johnstone, K.A., et al. 2009. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J.Am.Soc.Nephrol., 20, (5) 1123-1131 available from: PM:19389850 Bean, S.A., Bednarek, F.J., & Primack, W.A. 1995. Aggressive respiratory support and unilateral nephrectomy for infants with severe perinatal autosomal recessive polycystic kidney disease. J Pediatr, 127, (2) 311-313 available from: PM:7636663 Bergmann, C., Senderek, J., Windelen, E., et al. 2005. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int., 67, (3) 829-848 Decramer, S., Parant, O., Beaufils, S., et al. 2007. Anomalies of the TCF2 gene are the main cause of fetal bilateral hyperechogenic kidneys. J Am Soc Nephrol, 18, (3) 923-933 available from: PM:17267738 Drozdz, D., Drozdz, M., Gretz, N., et al. 1998. Progression to end-stage renal disease in children with posterior urethral valves. Pediatr Nephrol, 12, (8) 630-636 available from: PM:9811384 Edghill, E.L., Bingham, C., Ellard, S., & Hattersley, A.T. 2006. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med.Genet., 43, (1) 84-90 available from: PM:15930087 Fain, P.R., McFann, K.K., Taylor, M.R., Tison, M., Johnson, A.M., Reed, B., & Schrier, R.W. 2005. Modifier genes play a significant role in the phenotypic expression of PKD1. Kidney Int., 67, (4) 1256-1267 available from: PM:15780078 Fick-Brosnahan, G.M., Tran, Z.V., Johnson, A.M., Strain, J.D., & Gabow, P.A. 2001. Progression of autosomal-dominant polycystic kidney disease in children. Kidney Int., 59, (5) 1654-1662 available from: PM:11318935 Godley, M.L., Desai, D., Yeung, et al. 2001. The relationship between early renal status, and the resolution of vesico-ureteric reflux and bladder function at 16 months. BJU.Int., 87, (6) 457-462 available from: PM:11298034 Gonzalez, C.C., Bitsori, M., & Tullus, K. 2007. Progression of chronic renal failure in children with dysplastic kidneys. Pediatr.Nephrol, 22, (7) 1014-1020 available from: PM:17380351 Guay-Woodford, L.M. & Desmond, R.A. 2003. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics, 111, (5 Pt 1) 1072-1080 available from: PM:12728091 Harris, P.C. & Rossetti, S. 2010. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat.Rev.Nephrol., 6, (4) 197-206 available from: PM:20177400 Hildebrandt, F., Attanasio, M., & Otto, E. 2009. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol, 20, (1) 23-35 available from: PM:19118152 Ismaili, K., Schurmans, T., Wissing, K.M., et al. 2001. Early prognostic factors of infants with chronic renal failure caused by renal dysplasia. Pediatr.Nephrol, 16, (3) 260-264 available from: PM:11322375 Jenkins, D., Bitner-Glindzicz, M., Malcolm, S., et al. 2005. De novo Uroplakin IIIa heterozygous mutations cause human renal adysplasia leading to severe kidney failure. J.Am.Soc.Nephrol., 16, (7) 2141-2149 available from: PM:15888565 Komura, M., Kanamori, Y., Sugiyama, M., et al. 2008. Antenatally diagnosed giant multicystic dysplastic kidney resected during the neonatal period. J.Pediatr.Surg., 43, (11) 2118-2120 available from: PM:18970954 Liapis, H. & Winyard, P. J. D. 2007, "Cystic diseases and developmental kidney defects," In Heptinstall's Pathology of the KidneyJennette JC,. Olson JL, Schwartz MM, Silva FG, 6 ed. vol. 2 J. C. Jennette et al., eds., Philadephia, USA: Lippincott Williams and Wilkins, pp. 1257-1306. McPherson, E., Carey, J., Kramer, A., Hall, J.G., Pauli, R.M., Schimke, R.N., & Tasin, M.H. 1987. Dominantly inherited renal adysplasia. Am.J.Med.Genet., 26, 863-872 Murthi,G.V.S., AZMY,A.F., & Wilkinson,A.G. 2001. Management of simple renal cysts in children. J.R.Coll.Surg.Edinb., 46, 205-207 Narchi, H. 2005a. Risk of hypertension with multicystic kidney disease: a systematic review. Arch.Dis.Child, 90, (9) 921-924 available from: PM:15871982 Narchi, H. 2005b. Risk of Wilms' tumour with multicystic kidney disease: a systematic review. Arch.Dis.Child, 90, (2) 147-149 available from: PM:15665166 Ong, A.C. & Wheatley, D.N. 2003. Polycystic kidney disease–the ciliary connection. Lancet, 361, (9359) 774-776 available from: PM:12620752 Rickwood, A.M., Anderson, P.A., & Williams, M.P. 1992. Multicystic renal dysplasia detected by prenatal ultrasonography. Natural history and results of conservative management. Br.J Urol., 69, (5) 538-540 Rottenberg, G.T., Gordon, I., & de Bruyn, R. 1997. The natural history of the multicystic dysplastic kidney in children. Br.J Radiol., 70, (832) 347-350 available from: PM:9166069 Roy, S., Dillon, M.J., Trompeter, R.S., & Barratt, T.M. 1997. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol, 11, (3) 302-306 available from: PM:9203177 Rudnik-Schoneborn, S., John, U., Deget, F., et al. 1998. Clinical features of unilateral multicystic renal dysplasia in children. Eur.J Pediatr, 157, (8) 666-672 available from: PM:9727853 Van Dyck, M., Sidler, S., & Proesmans, W. 1998. Chronic renal failure in infants: effect of strict conservative treatment on growth. Eur.J Pediatr, 157, (9) 759-762 available from: PM:9776537 Wang, S., Zhang, J., Nauli, S.M., Li, X., Starremans, P.G., Luo, Y., Roberts, K.A., & Zhou, J. 2007. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol.Cell Biol., 27, (8) 3241-3252 Weber, S., Moriniere, V., Knuppel, T., et al. 2006. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol, 17, (10) 2864-2870 available from: PM:16971658 Weber, S., Taylor, J.C., Winyard, P., et al. 2008. SIX2 and BMP4 mutations associate with anomalous kidney development. J.Am.Soc.Nephrol., 19, (5) 891-903 available from: PM:18305125 Wilson, P.D. 2004. Polycystic kidney disease: new understanding in the pathogenesis. Int.J.Biochem.Cell Biol., 36, (10) 1868-1873 available from: PM:15203099 Winyard, P. & Chitty, L. 2001. Dysplastic and polycystic kidneys: diagnosis, associations and management. Prenat.Diagn., 21, (11) 924-935 available from: PM:11746145 Winyard, P. & Chitty, L.S. 2008. Dysplastic kidneys. Semin.Fetal Neonatal Med., 13, (3) 142-151 available from: PM:18065301 Winyard, P. J. D. & Jenkins, D. Putative roles of cilia in polycystic kidney disease. BBA - Molecular Basis of Disease . 2011. Ref Type: In Press Wuhl, E., Trivelli, A., Picca, S., et al. 2009. Strict blood-pressure control and progression of renal failure in children. N.Engl.J Med., 361, (17) 1639-1650 available from: PM:19846849 Yang, S.P., Woolf, A.S., Quinn, F., & Winyard, P.J.D. 2001. Deregulation of renal transforming growth factor-b1 after experimental short-term ureteric obstruction in fetal sheep. Am.J.Pathol., 159, 109-117