Cystic Kidney Disease
Paul JD Winyard and Karen L Price
Nephro-Urology Unit,
UCL Institute of Child Health
30 Guilford St, London, WC1N 1EH, UK
Corresponding author: [email protected]
→ Enlace a la versión en español

Paul JD Winyard and Karen L Price
Nephro-Urology Unit,
UCL Institute of Child Health
30 Guilford St, London, WC1N 1EH, UK
Corresponding author: [email protected]
→ Enlace a la versión en español
Overview
By definition, cystic kidneys contain cysts in place of part or all of the normal renal tissue, but there are many types and marked clinical heterogeneity. For classification, cystic kidneys can be broken down into genetic or non-genetic causes but new technologies are rapidly reducing the proportion in the latter group and it is clear that many apparently sporadic cases do have underlying mutations. Polycystic kidney disease is the classical genetic form and it is split into autosomal dominant (ADPKD) and recessive (ARPKD) inheritance. ADPKD mutations are much commoner (1 in 6-800) than ARPKD (1 in 5,000 mutation frequency, equating to 1 in 20,000 disease) but it is the latter which is more likely to cause problems with antenatal and early presentation with chronic kidney disease during childhood. The other major inherited forms are the recently named ‘ciliopathies’ where defects in development or function of primary cilia lead to kidney cysts, laterality defects and diverse pathologies. Examples include Bardet-Biedl syndromes, Nephronopthisis and there is a move to include PKD itself in this group too. Sporadic cystic kidneys fall into the multicystic/cystic dysplastic spectrum where the cysts are often part of a wider developmental defect in the kidneys and urinary tract. These are sometimes termed congenital anomalies of the kidney and urinary tract (CAKUT).
Antenatal diagnosis
Cystic kidneys are readily detected by ultrasound, so they are increasingly being diagnosed on antenatal scan (Winyard and Chitty 2008). There is a catch, however, in that the early stages of any cyst development or the multiple small cysts of ARPKD are below the resolution of ultrasound. Hence, there may be non-specific appearances rather than frank cysts, including one or more of increased size, bright echotexture and loss of cortico-medullary differentiation. ADPKD can also present like this in the newborn period but more classically exhibits a few large cysts which multiply and expand over time. Multicystic dysplastic kidneys also have moderate to large cysts from presentation but there is little normal renal tissue between them.
ADPKD
ADPKD accounts for around 5-10% of adult patients on dialysis or with a renal transplant in the developed world, but it rarely causes problems in childhood (Fick-Brosnahan et al. 2001). Ninety % of patients have an affected parent, and diagnosis can virtually be confirmed by finding cysts therein. Renal cysts develop progressively over many decades, however, so one has to be careful about negative results if the parents are young. A useful guide is that around 95% of affected individuals will have cysts by the time they reach 30 (Liapis and Winyard 2007). Renal complications include stones, infections, flank pain and gross haematuria, but there is also a risk of hypertension, liver and pancreatic cysts, and cerebral aneurysms.
Mutations in PKD1 or PKD2 cause over 98% of ADPKD (Harris and Rossetti 2010). A third locus (PKD3) has been postulated but never proven. PKD1 mutations have earlier onset of symptoms compared to PKD2 but both genes exhibit extensive allelic heterogeneity and variable clinical manifestations, perhaps attributable to genetic background and environmental differences which modify pathogenesis (Fain et al. 2005). PKD1 is located on chromosome 16p13.3 and encodes a 460 kDa protein, polycystin 1. PKD2 maps to 4q13-q23 and encodes polycystin 2, a calcium channel which interacts with its larger counterpart in a transmembrane polycystin complex in the plasma membrane. Localisation includes cilia but both polycystins are also found in many other sub-cellular sites including basal and lateral cell junctions (Wilson 2004)
ARPKD
ARPKD is much less common than ADPKD, but more likely to present before birth or in childhood so it was often known as ‘childhood PKD’ before its inheritance and genetics were defined. This confusing term should not be used since there is now plenty of the dominant disease diagnosed in childhood because of family genetic testing. ARPKD typically presents antenatally because of bright, enlarged kidneys and there is a high chance of perinatal death from immature lung development if there is severe oligo- or anhydramnios before 24 weeks gestation (Winyard and Chitty 2001). Postnatally, affected children may be diagnosed serendipitously because of enlarged kidneys but they are more likely to present with symptoms and signs of failing renal function. Of note, hypertension is common in ARPKD and can require multiple medications to control. Hepatic fibrosis can also become an issue as the child gets older. Around 85% of ARPKD patients survive the perinatal period, and historical US figures suggest 78% at 1 year and 75% survival at 5 years (Guay-Woodford and Desmond 2003). Parallel studies from the United Kingdom suggest actuarial renal survival of 86% at 1 year and 67% at 15 years for those patients that survive the first month (Roy et al. 1997).
Mutations in the polycystic kidney and hepatic disease 1 gene (PKHD1) cause ARPKD; this is located on chromosome 6p21 and encodes a protein named fibrocystin (or polyductin by different authors). There are a large number of known mutations spread throughout the gene, and the majority of patients are compound heterozygotes. There is a high risk of fetal presentation and neonatal death if the child carries two truncating mutations (Bergmann et al. 2005). Fibrocystin localises strongly to the primary cilium of renal epithelial cells, and there is evidence that this may also form part of the wider polycystin complex (Wang et al. 2007). Cysts only derive from the collecting ducts in ARPKD, but can come from all nephron segments in ADPKD (although 85-90% still arise from collecting ducts!).
Simple cysts
Simple kidney cysts are usually solitary and have no functional consequence. Their aetiology is unknown, but they are not inherited and cysts become commoner as people get older; indeed it is estimated that almost a third of those above 70 have at least one simple cyst. Children rarely have simple cysts, but there are case reports of isolated cysts with normal renal function, blameless ultrasound of the rest of the kidney and urinary tract and no evidence of hypertension (Murthi et al. 2001). Most cases are asymptomatic and only picked up by chance when abdominal/renal ultrasound is done for other purposes. Pain can occur secondary to the cysts, however, and cysts occasionally become infected or start to bleed. Standard adult therapy for these complications includes aspiration and injection of sclerosant but these must be undertaken with caution in children since the cysts may be the first manifestation of polycystic kidney disease, where it is well-recognised that these techniques do not prevent further cyst formation.
Cystic dysplasia and multicystic dysplastic kidneys
Dysplasia implies abnormal development and/or tissue components, which immediately highlights a difference with PKD because cysts in the latter are generated from normal parts of the nephron whereas there is reduced through zero ‘normal tissues’ within dysplastic organs. Instead, dysplastic kidneys contain poorly branched/differentiated nephrons and collecting ducts with increased stroma and smooth muscle (Winyard & Chitty 2008). Occasionally, there are also metaplastic areas of cartilage. Dysplastic kidneys are common with unilateral maldevelopment occurring in 1 in 1000, and bilateral in 1 in 5000, although it is only a subset of these that contain significant cysts. Cystic dysplastic kidneys represent one end of this spectrum, often containing areas with relatively normal nephrons adjacent to dysplastic regions with small to moderate cysts. Multicystic dysplastic kidneys are the other end, comprising just a collection of cysts with no functioning renal tissue. They are often connected to atretic ureters, and many of the molecular features of dysplasia can be replicated by obstruction in experimental animals but it is likely that genetic influences and environmental factors also play a part in pathogenesis (Yang et al. 2001).
Antenatal ultrasound often picks up dysplastic kidneys at the routine 20-week scan, usually as large bright kidneys, with or without cystic spaces. They may also be small, but there is ascertainment bias for larger ones, as they are easier to visualise and smaller kidneys may be masked by (or attributed to) technical difficulties. Multicystic dysplastic kidneys have characteristic appearances of multiple thin-walled non-connecting cysts in an enlarged kidney with an irregular non-reniform outline. They are sometimes difficult to distinguish from severe hydronephrosis with calyceal dilatation but the lack of a normally-situated renal pelvis and absent arterial flow using colour Doppler ultrasound can be diagnostic. Many dysplastic kidneys involute as the fetus / child grows (Rickwood et al. 1992;Rottenberg et al. 1997) and they can disappear before or after birth. The critical factor for longterm renal function is whether the contralateral kidney undergoes compensatory hypertrophy (Rudnik-Schoneborn et al. 1998).
Most dysplastic kidneys appear sporadic, although a family history of renal or urinary tract malformations was historically elicited in 10-15% of cases (McPherson et al. 1987). The commonest genetic cause is mutations of the TCF2 gene which encodes the protein hepatocyte nuclear factor (HNF) 1β. This was initially reported with cystic kidneys and maturity-onset diabetes of the young, but the malformation spectrum now includes cystic dysplastic, hypoplastic, polycystic and glomerulocystic kidneys, as well as solitary kidneys where the abnormal one may have involuted (Edghill et al. 2006). In recent analyses of selected populations, TCF2 mutations were reported in 29% of a series of 62 patients with antenatally diagnosed bright kidneys (Decramer et al. 2007) and 23% at our specialist referral hospital (Adalat et al. 2009). Mutations have also been reported with dysplastic kidneys in many other genes over the last few years including PAX-2 (renal-coloboma syndrome), Uroplakins, BMP4, SIX-1 and 2, EYA1 (Branchio-oto-renal syndrome), and SALL1 (Townes-Brocks syndrome) (Jenkins et al. 2005;Weber et al. 2006;Weber et al. 2008). There are also several inherited glomerulocystic diseases which combine variable elements of dysplasia (from mild hypoplasia through frank cystic dysplasia) with clear cysts arising from Bowman’s capsule. These can sometimes be mistaken for ADPKD, but many of the syndromic cases such as Oro-facial digital syndrome have classical abnormalities in other organs which point towards the true diagnosis.
The likelihood of developing chronic renal failure in patients with bilateral dysplasia is difficult to predict before birth, but there is a clear correlation between early childhood renal function and long-term outcome (Ismaili et al. 2001;Van Dyck et al. 1998). It is worth noting, however, that the normal postnatal renal maturation can be delayed by several years in dysplastic kidneys (Gonzalez et al. 2007). Prognosis of dysplasia associated with lower urinary tract malformations cannot be estimated accurately until the abnormalities are corrected (Drozdz et al. 1998;Godley et al. 2001).
Anecdotal reports suggested that dysplastic kidneys risk developing hypertension and tumours, but recent systematic reviews have suggested that these complications are very uncommon (Narchi 2005a;Narchi 2005b), and there is no evidence-based reason for elective nephrectomy unless there are complications such as frequent urinary tract infections.
Ciliopathy-based diseases
There are an increasing number of conditions that are classified as ciliopathies because the mutated genes involved encode proteins that localise to the primary cilium (Hildebrandt et al. 2009). These are membrane-enclosed finger-like organelles that project out from the cell, usually on the apical surface or facing into a lumen, duct or airway (Ong and Wheatley 2003). Again, the terminology is confusing because most textbooks include discussion of diseases such as Kartagener’s syndrome and say that this results from primary ciliary dyskinesia, but the defect is not in primary cilia at all! The primary defect there is in motile cilia which have the classical ‘9+2’ arrangement of structural microtubules necessary for coordinated beating to clear respiratory secretions. True primary cilia have a ‘9+0’ arrangement lacking the central microtubules and these have multiple roles in mechanosensation, photoreception and chemosensation. It is the first of these which links the ciliopathies to cystic kidney diseases: physiological fluid flow across the apical surface of renal cells bends the cilia and induces calcium influx, and this is perturbed when the ciliopathy genes are mutated. Downstream effects include changes in cell differentiation and polarity, with perturbation of iconic pathways such as Hedgehog and Wnt signalling (Winyard and Jenkins 2011).
The PKD genes are expressed in the primary cilium and clearly have massive cyst formation and destruction of normal tissues, but many of the other conditions on the ciliopathy list have their major abnormalities in other organ systems such as the heart, eye or nervous system; hence, they will not be discussed further here but there are several excellent recent reviews (Waters and Beales 2011). For reference, the current list includes Bardet-Biedl Syndrome, Nephronophthisis, Alstrom Syndrome, Orofaciodigital Syndrome, Senior-Loken Syndrome and Usher Syndrome.
Treatment of cystic kidney diseases
There are not currently any specific treatments that target the development of cysts. Hence, the mainstay of treatment for all of these conditions is supportive care to regulate blood pressure and manage complications of chronic kidney disease (CKD). Blockade of the renin-angiotensin pathway using ACE inhibitors and receptor blockers is routine therapy in most CKD, particularly when there is significant proteinuria, and has recently been shown to slow renal deterioration in children (Wuhl et al. 2009). Surgery is only really useful to correct urological abnormalities, and cyst deroofing or aspiration is rarely used since cysts rapidly re-accumulate. Nephrectomy is occasionally warranted before transplant if the kidneys are massive, but most organs tend to fibrose and become smaller as end-stage is reached. There are rare case reports of emergency removal of cystic organs that splint the diaphragm and/or interfere with feeding in the neonatal period (Bean et al. 1995;Komura et al. 2008)
Experimental animal work on PKD has uncovered a raft of new potential treatments for some conditions in the past 5 years. These include modulation of the vasopressin system using tolavaptan/similar agents or increased water intake; blockade of mTOR pathways using rapamycin-like drugs, and somatostatin. The updated list of trials can be viewed at http://www.pkdcure.org/Research/ClinicalTrials/ActiveRecruiting.aspx It is hoped that one or more of these may be successful and that these treatments can then be trialled for other paediatric cystic diseases in future.
Figure
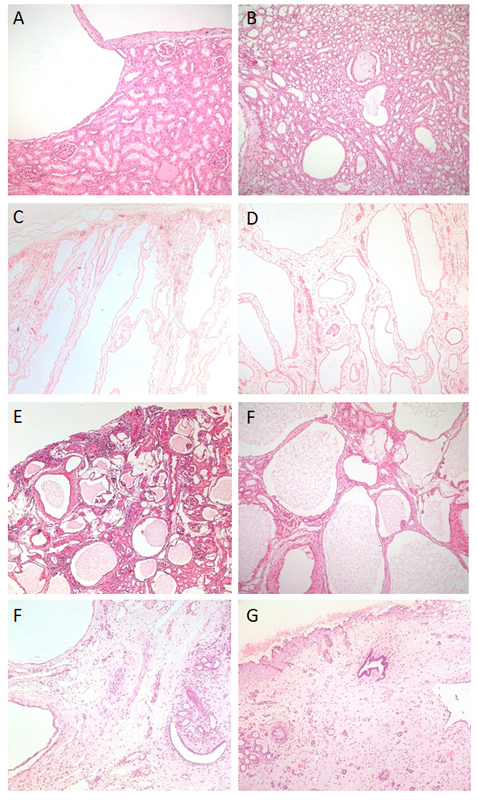
Haematoxylin and Eosin-stained sections from paediatric cystic kidneys. Views of the cortex are shown in the left column and medulla in the right. A and B) Autosomal Dominant PKD with a few cysts in both cortex and medulla arising from all nephron segments. This was from an early stage, hence the well-preserved architecture of the normal surrounding glomeruli, tubules and collecting ducts. C and D) Autosomal Recessive PKD with numerous elongated cysts running through the cortex and medulla; note that all of the glomeruli appear to have a normal, undilated Bowmans space since cysts only arise from collecting ducts in this condition. E and F) Glomerulocystic kidney diseases with marked dysplasia; note the significant disruption of renal differentiation and the glomerular tufts visible in virtually every cyst. G and H) Cystic dysplastic kidneys display virtually no normal renal structures, instead there are large areas of poorly differentiated stroma, a few irregular dilated tubules and numerous cysts.
References
Adalat, S., Woolf, A.S., Johnstone, K.A., Wirsing, A., Harries, L.W., Long, D.A., Hennekam, R.C., Ledermann, S.E., Rees, L., van't, H.W., Marks, S.D., Trompeter, R.S., Tullus, K., Winyard, P.J., Cansick, J., Mushtaq, I., Dhillon, H.K., Bingham, C., Edghill, E.L., Shroff, R., Stanescu, H., Ryffel, G.U., Ellard, S., & Bockenhauer, D. 2009. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J.Am.Soc.Nephrol., 20, (5) 1123-1131 available from: PM:19389850
Bean, S.A., Bednarek, F.J., & Primack, W.A. 1995. Aggressive respiratory support and unilateral nephrectomy for infants with severe perinatal autosomal recessive polycystic kidney disease. J Pediatr, 127, (2) 311-313 available from: PM:7636663
Bergmann, C., Senderek, J., Windelen, E., Kupper, F., Middeldorf, I., Schneider, F., Dornia, C., Rudnik-Schoneborn, S., Konrad, M., Schmitt, C.P., Seeman, T., Neuhaus, T.J., Vester, U., Kirfel, J., Buttner, R., & Zerres, K. 2005. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int., 67, (3) 829-848
Decramer, S., Parant, O., Beaufils, S., Clauin, S., Guillou, C., Kessler, S., Aziza, J., Bandin, F., Schanstra, J.P., & Bellanne-Chantelot, C. 2007. Anomalies of the TCF2 gene are the main cause of fetal bilateral hyperechogenic kidneys. J Am Soc Nephrol, 18, (3) 923-933 available from: PM:17267738
Drozdz, D., Drozdz, M., Gretz, N., Mohring, K., Mehls, O., & Scharer, K. 1998. Progression to end-stage renal disease in children with posterior urethral valves. Pediatr Nephrol, 12, (8) 630-636 available from: PM:9811384
Edghill, E.L., Bingham, C., Ellard, S., & Hattersley, A.T. 2006. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med.Genet., 43, (1) 84-90 available from: PM:15930087
Fain, P.R., McFann, K.K., Taylor, M.R., Tison, M., Johnson, A.M., Reed, B., & Schrier, R.W. 2005. Modifier genes play a significant role in the phenotypic expression of PKD1. Kidney Int., 67, (4) 1256-1267 available from: PM:15780078
Fick-Brosnahan, G.M., Tran, Z.V., Johnson, A.M., Strain, J.D., & Gabow, P.A. 2001. Progression of autosomal-dominant polycystic kidney disease in children. Kidney Int., 59, (5) 1654-1662 available from: PM:11318935
Godley, M.L., Desai, D., Yeung, C.K., Dhillon, H.K., Duffy, P.G., & Ransley, P.G. 2001. The relationship between early renal status, and the resolution of vesico-ureteric reflux and bladder function at 16 months. BJU.Int., 87, (6) 457-462 available from: PM:11298034
Gonzalez, C.C., Bitsori, M., & Tullus, K. 2007. Progression of chronic renal failure in children with dysplastic kidneys. Pediatr.Nephrol, 22, (7) 1014-1020 available from: PM:17380351
Guay-Woodford, L.M. & Desmond, R.A. 2003. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics, 111, (5 Pt 1) 1072-1080 available from: PM:12728091
Harris, P.C. & Rossetti, S. 2010. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat.Rev.Nephrol., 6, (4) 197-206 available from: PM:20177400
Hildebrandt, F., Attanasio, M., & Otto, E. 2009. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol, 20, (1) 23-35 available from: PM:19118152
Ismaili, K., Schurmans, T., Wissing, K.M., Hall, M., Van Aelst, C., & Janssen, F. 2001. Early prognostic factors of infants with chronic renal failure caused by renal dysplasia. Pediatr.Nephrol, 16, (3) 260-264 available from: PM:11322375
Jenkins, D., Bitner-Glindzicz, M., Malcolm, S., Hu, C.C., Allison, J., Winyard, P.J., Gullett, A.M., Thomas, D.F., Belk, R.A., Feather, S.A., Sun, T.T., & Woolf, A.S. 2005. De novo Uroplakin IIIa heterozygous mutations cause human renal adysplasia leading to severe kidney failure. J.Am.Soc.Nephrol., 16, (7) 2141-2149 available from: PM:15888565
Komura, M., Kanamori, Y., Sugiyama, M., Nakahara, S., Kawashima, H., Hatanaka, A., Takazawa, Y., Goishi, K., Igarashi, T., & Iwanaka, T. 2008. Antenatally diagnosed giant multicystic dysplastic kidney resected during the neonatal period. J.Pediatr.Surg., 43, (11) 2118-2120 available from: PM:18970954
Liapis, H. & Winyard, P. J. D. 2007, "Cystic diseases and developmental kidney defects," In Heptinstall's Pathology of the KidneyJennette JC,. Olson JL, Schwartz MM, Silva FG, 6 ed. vol. 2 J. C. Jennette et al., eds., Philadephia, USA: Lippincott Williams and Wilkins, pp. 1257-1306.
McPherson, E., Carey, J., Kramer, A., Hall, J.G., Pauli, R.M., Schimke, R.N., & Tasin, M.H. 1987. Dominantly inherited renal adysplasia. Am.J.Med.Genet., 26, 863-872
Murthi,G.V.S., AZMY,A.F., & Wilkinson,A.G. 2001. Management of simple renal cysts in children. J.R.Coll.Surg.Edinb., 46, 205-207
Narchi, H. 2005a. Risk of hypertension with multicystic kidney disease: a systematic review. Arch.Dis.Child, 90, (9) 921-924 available from: PM:15871982
Narchi, H. 2005b. Risk of Wilms' tumour with multicystic kidney disease: a systematic review. Arch.Dis.Child, 90, (2) 147-149 available from: PM:15665166
Ong, A.C. & Wheatley, D.N. 2003. Polycystic kidney disease--the ciliary connection. Lancet, 361, (9359) 774-776 available from: PM:12620752
Rickwood, A.M., Anderson, P.A., & Williams, M.P. 1992. Multicystic renal dysplasia detected by prenatal ultrasonography. Natural history and results of conservative management. Br.J Urol., 69, (5) 538-540
Rottenberg, G.T., Gordon, I., & de Bruyn, R. 1997. The natural history of the multicystic dysplastic kidney in children. Br.J Radiol., 70, (832) 347-350 available from: PM:9166069
Roy, S., Dillon, M.J., Trompeter, R.S., & Barratt, T.M. 1997. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol, 11, (3) 302-306 available from: PM:9203177
Rudnik-Schoneborn, S., John, U., Deget, F., Ehrich, J.H., Misselwitz, J., & Zerres, K. 1998. Clinical features of unilateral multicystic renal dysplasia in children. Eur.J Pediatr, 157, (8) 666-672 available from: PM:9727853
Van Dyck, M., Sidler, S., & Proesmans, W. 1998. Chronic renal failure in infants: effect of strict conservative treatment on growth. Eur.J Pediatr, 157, (9) 759-762 available from: PM:9776537
Wang, S., Zhang, J., Nauli, S.M., Li, X., Starremans, P.G., Luo, Y., Roberts, K.A., & Zhou, J. 2007. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol.Cell Biol., 27, (8) 3241-3252
Weber, S., Moriniere, V., Knuppel, T., Charbit, M., Dusek, J., Ghiggeri, G.M., Jankauskiene, A., Mir, S., Montini, G., Peco-Antic, A., Wuhl, E., Zurowska, A.M., Mehls, O., Antignac, C., Schaefer, F., & Salomon, R. 2006. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol, 17, (10) 2864-2870 available from: PM:16971658
Weber, S., Taylor, J.C., Winyard, P., Baker, K.F., Sullivan-Brown, J., Schild, R., Knuppel, T., Zurowska, A.M., Caldas-Alfonso, A., Litwin, M., Emre, S., Ghiggeri, G.M., Bakkaloglu, A., Mehls, O., Antignac, C., Network, E., Schaefer, F., & Burdine, R.D. 2008. SIX2 and BMP4 mutations associate with anomalous kidney development. J.Am.Soc.Nephrol., 19, (5) 891-903 available from: PM:18305125
Wilson, P.D. 2004. Polycystic kidney disease: new understanding in the pathogenesis. Int.J.Biochem.Cell Biol., 36, (10) 1868-1873 available from: PM:15203099
Winyard, P. & Chitty, L. 2001. Dysplastic and polycystic kidneys: diagnosis, associations and management. Prenat.Diagn., 21, (11) 924-935 available from: PM:11746145
Winyard, P. & Chitty, L.S. 2008. Dysplastic kidneys. Semin.Fetal Neonatal Med., 13, (3) 142-151 available from: PM:18065301
Winyard, P. J. D. & Jenkins, D. Putative roles of cilia in polycystic kidney disease. BBA - Molecular Basis of Disease . 2011. Ref Type: In Press
Wuhl, E., Trivelli, A., Picca, S., Litwin, M., Peco-Antic, A., Zurowska, A., Testa, S., Jankauskiene, A., Emre, S., Caldas-Afonso, A., Anarat, A., Niaudet, P., Mir, S., Bakkaloglu, A., Enke, B., Montini, G., Wingen, A.M., Sallay, P., Jeck, N., Berg, U., Caliskan, S., Wygoda, S., Hohbach-Hohenfellner, K., Dusek, J., Urasinski, T., Arbeiter, K., Neuhaus, T., Gellermann, J., Drozdz, D., Fischbach, M., Moller, K., Wigger, M., Peruzzi, L., Mehls, O., & Schaefer, F. 2009. Strict blood-pressure control and progression of renal failure in children. N.Engl.J Med., 361, (17) 1639-1650 available from: PM:19846849
Yang, S.P., Woolf, A.S., Quinn, F., & Winyard, P.J.D. 2001. Deregulation of renal transforming growth factor-b1 after experimental short-term ureteric obstruction in fetal sheep. Am.J.Pathol., 159, 109-117
 |
 |
 |
 |
 |
 |
 |
 |
