Disorders of sex development: the issues for a urologist
John M Hutson
→ Enlace a la versión en español

John M Hutson
→ Enlace a la versión en español
Urology Department
Royal Children’s Hospital
Parkville, Victoria 3052
AUSTRALIA
Introduction
Disorders of sex development (DSD) are a complex group of anomalies affecting the internal and external genitalia. Because they tend to be both complicated and rare, management is best done by a regional team of experts, who can maintain their expertise by frequent exposure. The paediatric urologist is an essential part of this team, and can contribute to understanding the normal and abnormal embryology, as well as the genital anatomy at presentation, particularly in the physical diagnosis of the disorder. This chapter outlines these various areas along with the principles of surgical management. For a comprehensive treatise on DSD the reader is referred to our book on DSD (Hutson, Warne and Grover, 2012).
Normal Embryology
During early embryonic development the urogenital ridge forms from the intermediate mesoderm as a longitudinal protrusion on the posterior wall of the pleuroperitoneal cavity (1,2). Originally a few tubules form at the end of the urogenital ridge and then almost immediately regress as the transient pronephros. By the time the diaphragm forms to create the abdominal cavity the mesonephros or middle kidney has developed and has taken over the lower half of the pronephric duct, so it becomes the mesonephric or Wolffian duct. On the anteromedial surface of the mesonephros the mesenchyme condenses to form the ambisexual gonad, into which the primitive germ cells migrate from the caudal yolk sac stalk in the region of the allantois. Near the cranial pole of the gonad the peritoneal surface over the lateral free edge of the urogenital ridge evaginates to form the paramesonephric or Müllerian duct, which then elongates caudally to the cloaca following the Wolffian duct. Caudally the urogenital ridges roll medially so that their free edges , each with a contained Müllerian duct,
come into apposition with each other, allowing the ducts to fuse, to create the uterovaginal primordium. The caudal end of the fused ducts, which is a solid cord, migrates to the anterior cloaca between the Wolffian ducts to stimulate proliferation of the bulb endoderm to form the sinovaginal bulb. In females further proliferation of this endoderm caudally extends the uterovaginal primordium to the outside, immediately behind the primitive urethra.
Sexual development of the internal genitalia beginsat 7-8 weeks’ gestation.SRY expression in males triggers a cascade of differentiation in the ambisexual gonad to form a testis (3). Early production of anti-Müllerian hormone (AMH; also called Müllerian Inhibiting Substance, MIS) causes regression of the Müllerian ducts, while testosterone from the Leydig cells is secreted as an exocrine hormone down the Wolffian ducts to trigger their development into epididymis cranially, vas deferens caudally and budding of the distal Wolffian duct to produce the seminal vesicle (4). Testosterone also enters the blood as an endocrine hormone to reach the external genitalia, where it is converted by an enzyme, 5alpha-reductase, into dihydrotestosterone (DHT) (5). DHT binds 5-10 times more tightly to androgen receptors than testosterone itself, thereby functioning as a concentrating mechanism for androgenic action at a time when the developing testis is still tiny and the blood level of testosterone is low.
Circulating androgens, via DHT, masculinise the external genitalia at 8-12 weeks. The genital tubercle enlarges into a penis, while the endodermal urethral plate canalises to form a urethra that extends initially to the future coronal groove (~ 12 weeks) and eventually to the tip of the glans (~15-20 weeks). The inner and outer genital folds grow over the urethral plate to form the periurethral tissues and scrotum, and the uterovaginal primordium is stimulated to regress.
In addition, circulating androgens cause regression of the cranial suspensory ligament of the gonad.
The developing Leydig cells also produce insulin-like hormone 3 (Insl3), which stimulates the caudal suspensory ligament of the urogenital ridge, known as the genito-inguinal ligament or “gubernaculum” (6-8). The gubernaculum ends in the anterior abdominal wall just lateral to the developing Rectus abdominis muscle. Its caudal end enlarges in the male in response to Insl3. In addition, its attachment to the urogenital ridge and lower pole of the gonad (known as the cord) remains short and thick, thereby anchoring the testis to the future inguinal canal as the fetal abdominal cavity enlarges with growth (9). This anchoring of the fetal testis between 8 and 15 weeks of gestation causes the first or transabdominal phase of descent.
At about 25 weeks’ gestation the fetal testis starts to descend as part of the inguino-scrotal phase of descent.Migration to the scrotum requires the gubernaculum to elongate beyond the external inguinal ring, across the pubic region and into the scrotum, probably controlled by the genitofemoral nerve. The latter is regulated by androgens, which stimulate release of a neurotransmitter, calcitonin gene-related peptide (CGRP), from the sensory branches of the nerve to provide a chemical gradient for the gubernaculum to follow. After descent is complete CGRP also regulates obliteration of the proximal processus vaginalis to prevent inguinal hernia.
In females genital development proceeds initially without hormones.Absence of AMH allows the Müllerian ducts to differentiate into Fallopian tubes, uterus and upper vagina, while lack of testosterone not only allows the external genitalia to retain their female shape but also allows
vaginal development and canalisation. Without androgens the urethral plate of the future clitoris undergoes apoptosis, thereby shortening the ventral surface to produce the natural hair-pin typical of mature clitoral anatomy (10). The regressing mesonephros leaves the gonads on a mesentery, which in females becomes a substantial structure as the mesovarium and the rest of the urogenital ridge develops into the broad-ligament, to anchor the female genital tract in the pelvis (11).Absence of Insl3 in the developing ovary allows the female gubernaculum to elongate in proportion to fetal growth, to eventually become the ligament of the ovary and the round ligament.
Abnormal Embryology
Failure of testicular differentiation interferes with the hormones that control sexual differentiation (12). Where the anomaly affects the amount of testicular hormones, the degree of masculinisation, both internally and externally, is proportional to the functional levels of testosterone, AMH and Insl3 that are produced. The amount of effective androgenic signalling (via DHT) can be determined from the external genitalia, which are in effect a “bioassay” for androgen (13).
In complex chromosomal anomalies, such as aneuploidy and mosaicism, there may be asymmetrical testicular development. In sex chromosome mosaicism the gonadal differentiation is determined by the number of cells in the ambisexual gonad that contain a Y chromosome, with normal SRY expression. It is common for the percentage of cells containing a Y chromosome to vary in each urogenital ridge, so that ‘testicular’ development may occur only on one side. Because the internal genital ducts respond to exocrine hormones from the ipsilateral testis the Wolffian duct will be preserved on the side of a testis, while the Müllerian duct will be absent on this same side. As the testes control their own descent (via Insl3, AMH and testosterone), ductal asymmetry is paired with asymmetrical testicular descent in newborns with mixed gonadal dysgenesis (45 X/46XY, with a streak gonad on one side and a testis on the other) and ovotesticular DSD (where there may be a testis on one side and ovary on the other, or various combinations of ovotestes).
Placental insufficiency can cause ambiguous genitalia. In the past these anomalies were invisible because of fetal death in utero or early postnatal deaths in extremely premature newborns. The effects of placental insufficiency are now seen in some surviving males born at 24-26 weeks’ gestation with severe intra-uterine growth retardation (IUGR), where early production of testicular hormones wasthought to be impeded by the lack of chorionic gonadotrophin (hCG) (5) from the abnormal placenta. The baby has IUGR and ambiguous genitalia with partially descended testes, but no other hormonal anomaly after birth. The growth failure prenatally means that many of these boys are tiny, and although catch-up growth occurs during childhood, some need growth hormone.
Abnormalities of the hypothalamic-pituitary-gonadal axis cause micropenis and cryptorchidism. In these disorders the infant has fully masculinised external genitalia (8-12 weeks) but failure of phallic growth between 15 and 40 weeks, leading to micropenis (14). This is because initial sexual differentiation is normal as the developing testis is stimulated by hCG from the placenta until the hypothalamic axis takes over at about 15 weeks of gestation. This leads to normal circulating androgens between 8 and 15 weeks, but deficient androgens thereafter. The two remaining features of masculine anatomy that develop after this, i.e. phallic enlargement and inguinoscrotal testicular descent, are the only things deranged.
Androgen function is disturbed by a number of specific enzymatic and receptor mutations.A classic example is the patient with complete androgen insensitivity syndrome (CAIS), where the androgen receptor is non-functional (19). External genital anatomy is that of a normal female apart from the presence of testes in the inguinal region (inside the inguinal canal or just outside the external inguinal ring), as the first phase of testicular descent is normal while the second phase is completely blocked. In addition, AMH function is normal so that the Müllerian ducts have regressed even though lower vaginal development is normal, as the uterovaginal primordium forms in the absence of androgens (20, 21).
Specific mutations in enzymes or receptors controlling the other testicular hormones cause isolated anatomical derangements. Insl3 synthesis or Insl3 receptor anomalies, for example, would produce undescended testes, but these are rare. However, endocrine disruptors in the maternal environment may account for some cases, as exogenous oestrogens suppress Insl3. Mutations in AMH signalling also lead to a rare syndrome, known as persisting Müllerian duct syndrome (PMDS) (15-17). In this abnormality external genital development is normal male but the testes are undescended and the Müllerian ducts persist as an infantile uterus, tubes and upper vagina (entering the posterior urethra near the verumontanum). The cause for cryptorchidism is debated, and many authors suggest that the retained uterus and broad ligaments prevent descent (18). The cord of the gubernaculum, however, is abnormally long, suggesting that AMH has some role in preventing gubernacular cord elongation, at least in humans, as this is not seen in rodents (15-17). An important corollary of the link between AMH and testicular descent is the fact that when transabdominal descent of the testis has occurred (so that the testis is palpable), we can confidently predict that the Müllerian duct (on that side) is regressed (13).
When the external genital anomaly is outside the narrow spectrum from normal female to normal male or the adjacent perineum is abnormal, the baby does not have a hormonal abnormality but instead has a complex morphological anomaly of the perineum (12). This range of anomalies is familiar to surgeons, and includes bladder and cloacal exstrophy, ectopic or absent scrotum or penis and many anorectal malformations with cutaneous, bulbar or vestibular fistulae(2).Clinical Examination and Rules for Diagnosis
The clinical examination of a baby with suspected DSD includes a careful history of the pregnancy and the family. The family history may reveal an inherited anomaly with an autosomal recessive or x-linked pattern, such as in androgen insensitivity. Possible prenatal factors in the mother or her environment (e.g. steroid ingestion, hormone-secreting tumours, placental dysfunction) may also interfere with sexual development in the fetus.
The physical examination of the genitalia requires a number of features to be determined, using some simple rules derived from our knowledge of normal and abnormal development (Table 1) (13). Clearly a general examination will include a search for other anomalies and possible syndromic features, as a number of malformation syndromes are associated with DSD (40). The genital examination should concentrate on the following:
(i) establish degree of masculinisation, which is easily done using the Prader Score from 0 to 5 (Prader scale: O: normal female appearance; 1-4: increasing masculinisation; 5: normal male appearance). This determines the amount of active androgen the baby was exposed to,as the degree of virilisation is directly proportional to the amount of androgen signalling. It also predicts the amount of lower vaginal regression, although some authors have disputed this (22).
(ii) determine the site, shape and contents of the urogenital opening. The labio-scrotal folds should be pulled open to examine the urogenital sinus. In boys with simple hypospadias, not only is the scrotum fused and contains two descended testes, but the urethral meatus is usually tiny. By contrast, in babies with a DSD causing androgen insufficiency the urogenital opening is a funnel-shaped opening. At the apex of this funnel it may be possible to see the urethral and vaginal confluence, which is marked by the hymen. The latter is easily recognised as its mucosa is a slightly paler bluish-pink colour in contrast to the bright red urethral mucosa. The pale blue colour of the hymen at birth is a result of stimulation by maternal oestrogens. If a hymen can be seen then we can deduce the presence of the lower vagina persisting from the vaginal plate.
(iii) determine the presence and site of the gonads, which, if present, are testes. Palpation of any testis will not only confirm its normal (or abnormal) consistency, but also will identify any epididymis or vas deferens. Where testes are not palpable, you should palpate the site of the external inguinal ring. If this is palpable as a triangular defect, then it is highly likely that there is a testis present inside the inguinal canal. By contrast, if the ring is completely closed the gonad is unlikely to be inside the canal, and it suggests that it is intra-abdominal, either as a very dysplastic or dysgenetic testis/streak gonad or is simply an ovary (which never descends as it lacks the testicular hormones that control descent). When an inguinal hernia contains an ovary in a girl, it has been pulled into a sliding hernia rather than ‘descended’ like a testis (Hutson & Kearsey, JPS in press).
(iv) determine the state of the internal ducts by rectal examination to palpate a cervix and/or arrange an ultrasonogram of the pelvis to see if ovaries and female ducts can be seen.
Surgical Management
The surgical treatment of the genital anomalies present in DSD requires a high degree of expertise. Not only does the surgery itself need considerable skill in reconstructive procedures but also because the outcome must be good enough to withstand critical adult appraisal half a lifetime later (23, 24)! After the discovery of congenital adrenal hyperplasia in 1953, there was an explosion in new techniques to treat DSD, but many procedures devised in the 1960’s,
1970’s and 1980’s had a relatively poor cosmetic and functional outcome when that cohort of patients reached adulthood (25). This led to the establishment of many patient support groups and some public backlash against amateurish surgical efforts, leading in some places in the world to a moratorium on genital surgery in infants (26, 27). However, this period of ‘trial and error’ is a natural part of the learning curve in surgery, so initial poor outcomes should be the stimulus for advancement, rather than abandonment of treatment.
Poor initial long-term outcomes triggered a radical reappraisal of management, with the establishment of regional centres of excellence to allow accumulation and maintenance of expertise and multidisciplinary skills. As in many aspects of paediatric surgery, the basic problem here was the long lag-time between the intervention (surgery in early childhood/infancy) and the functional, cosmetic and mental outcome measured in adulthood and the ability of these people to have a satisfying sex life. In the first 20-30 years of DSD surgery there was no major adult feedback except for anecdotal cases, but this has now been replaced with increasingly formalised follow-up studies, so that the best aspects of different approaches can be assimilated into a common management plan (28, 29, 39).
In our own institution the first long-term review was done in the 1980’s and this identified a number of important issues, as listed below.
i) children had nightmares about frequent (and often public) genital examination, in outpatient clinic, so we immediately altered practice to obtain informed consent (from the child, if possible) and appropriate privacy and discretion. Currently we only do a genital examination later in childhood if there is an indication.
(ii) Postoperative vaginal dilatations were traumatic (both physically and mentally) for children and parents, and were probably unnecessary if surgical healing was achieved without sepsis. We stopped this in the 1980’s, and developed more aggressive preventive measures against perineal wound infection (antibiotic cover, antiseptic ointment and salt baths).
(iii) regular follow-up by the operating surgeon provided invaluable moral support for children growing up as they had regular contact with the person who knew the most about their diagnosis and anatomy. This remains an important part of our current treatment plan.
(iv) early exposure (during childhood) to the adolescent gynaecologist was a great help for both parents and girls with DSD, as transition to adult care was facilitated by an unbroken chain. We currently introduce our female patients to the adolescent gynaecologist at about 9-11 years of age.
(v) open disclosure not only demystified the medical complexities of the diagnosis but also enabled DSD to be managed like any other chronic medical problem, rather than something secretive and shameful or a freak of nature. We started with disclosure to parents, and quickly appreciated that the sooner children begin to understand the underlying issues the less disturbed their personality development will be. Open discussion of the problem with age-specific vocabulary occurs throughout childhood, leading up to more formal explanations of the biology in late childhood or early adolescence.
(vi) ongoing counselling, both informal and formal was an absolute necessity, based on our increasing awareness of the brain as the most important organ of sexuality.
Timing
Despite the debate in many centres about avoiding irreversible surgery in infancy (see Garry Warne’s accompanying chapter) we continue to offer early intervention with fully-informed consent, if that is the parents’ wish. This is because long-term follow up shows that only about 12% of girls need further surgery in adolescence (presented at the International DSD meeting, Gent, Belgium, June 2015).
For female genitoplasty, we currently recommend surgery between 6 weeks and 6 months (30). This allows time for full diagnostic workup and counselling, without parents feeling that they are being pushed into it. The clitoromegaly recedes somewhat after the commencement of cortisone, and the secondary suppression of abnormal androgen production. By a few months little further shrinkage will occur and surgery is used to reduce the clitoris to the correct size. Beyond 9-12 months other factors coming into play, such as the baby crawling and walking, which risks trauma to the genitalia postoperatively if there is a fall (which is common!). In addition, the tissues are easy to work with at this age, and healing is rapid, as long as superficial infection and wound breakdown can be avoided by adequate perioperative perineal toilet.
For boys we usually begin reconstruction between 3 and 6 months, so further stages can be done (and hopefully completed) by 18 months of age, when permanent memories of admission to hospital and genital surgery may be an issue.
Female Genitoplasty
Surgical reconstruction, most commonly for babies with CAH, includes 3 basic principles (31-34).
![]()
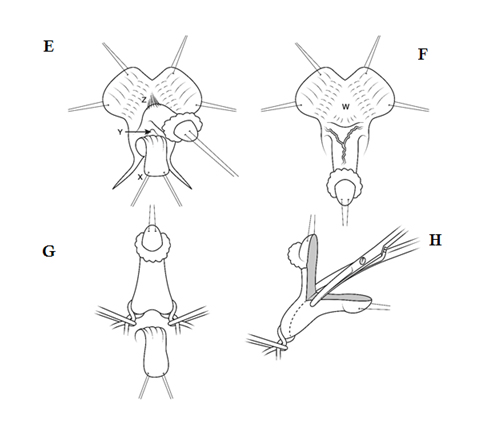
1. Clitoroplasty to reduce excess erectile tissue of the corpora cavernosa and reducing the size of the glans, while preserving the neurovascular supply so that subsequent sexual function is as close to normal as possible. There are numerous methods for clitoroplasty but we have continued to use a modification of the method first used in our department by Robert Fowler in the early 1970’s. This technique removes excess ventral erectile tissue and recreates the normal hairpin bend in the clitoris, and avoids any dissection on the dorsal surface near the neurovascular bundles (35) (Figure 1SHOULD THIS BE FIGURE C-I, INSTEAD??). Our results continue to support this approach against the alternative method of isolation of the neurovascular bundles to allow excision of the shaft of the phallus: the latter method has a greater risk of glans ischaemia or sensory loss which would seriously compromise adult sexual function (24, 29). The only good argument against our approach is the risk of enlargement of the corpora cavernosa in adolescence if the steroid suppression lapses.
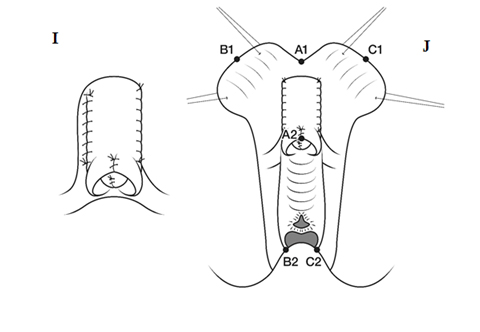
2. Creation of a normal-looking introitus with labia minora and labia majora. Although this seems like “trivial” cosmetic surgery, this is in fact one of the key steps in the surgery, as the external appearance is used by the patient herself (as well as everyone else, including potential male partners) as a measure of “normality”. The outer foreskin hood around the clitoris is partly used to create labia minora while the inner foreskin is left as a covering for the newly reconstructed clitoral glans (36).FIGURE I-L
3. The third aim of the genital reconstruction is a vaginoplasty, to provide an adequate opening in the introitus for the vagina (37, 38). This prevents urine pooling in the vagina if the common urogenital sinus is left intact, as well as allows normal tampon use in adolescence, with or without some minor patient-controlled dilatation at that time. There are many approaches to vaginoplasty, from simple Y-V plasty to Passerini operation (38), but we have found no need to do complex operations, as long as the V-flap is inserted high enough on the posterior vaginal wall to relieve the androgen-induced stenosis of the lower third of the vagina (29, 35). In addition, it is extremely rare in CAH patients for the confluence of the vagina with the urogenital sinus to encroach on the sphincter zone of the urethra. Only in the exceptional cases (with extreme virilisation, Prader V) has a Passerini-Glazel procedure or a pull-through been required.
Male Genitoplasty
Boys with ambiguous genitalia have severe hypospadias, and a staged reconstruction is helpful here. The principles of hypospadias repair hardly need emphasis in this chapter, as they are similar to that elsewhere in this text. However, there are a few minor differences worth a mention.
During the first procedure, endoscopy is helpful to identify and document the site and size of a retained lower vaginal remnant, as this may need to be excised if it is likely to store urine after distal urethroplasty. Also, laparoscopy may be required to document precisely the internal anatomy and/or excise a streak gonad. We have also used laparoscopy to detach the vaginal remnant from the urethra but without excision, so that the early hypospadias repair is not compromised by pooling urine in the vagina and secondary sepsis, but the options are left open in the future for gender change, should this occur.
Also during the first operation the chordee is released in the standard fashion, and orchidopexy and/or hernia repair can be performed. In addition a gonadal biopsy may be needed.
After a 6 month interval we would proceed to urethroplasty and correction of the penoscrotal transposition, along with repair of the bifid scrotum. Also,in some patients the connection between the vaginal remnant and urogenital sinus is so low it can be reached easily from the perineum by opening the anterior part of the perineal body. The vaginal remnant can be excised completely or disconnected and left in situ, and sometimes excision of the mucosa is sufficient. The latter is relatively simple, and decreases the risk of inadvertent damage to the rectum.
Table 1 Rules for Diagnosis
Table 1 Rules for Diagnosis
Rule No |
|
|
|
|
|
1 |
Testes descend, ovaries don’t |
A palpable gonad is a testis |
|
|
|
2 |
Testis descent + Mullerian |
A palpable gonad means ipsilateral Müllerian |
|
|
|
3 |
Uterus present = Sertoli cells |
Persisting Müllerian ducts onlyoccurwhen testis is absent or dysgenetic (or rarePMDS) |
|
|
|
4 |
Internal ducts mirror ipsilateral gonad |
MD/WD controlled by ipsilateral exocrine |
|
|
|
5 |
Circulatory (adrenal) androgens cannotmasculinise Wolffian duct |
Adrenal androgen levels (in CAH) too low at 8 weeks and no exocrine secretion down WD |
|
|
|
6 |
External genitaliamasculinise proportional to androgen exposure |
Clitoral enlargement (erectile tissue, not just foreskin)only caused by androgen |
|
|
|
7 |
External masculine development inversely proportional to lower vaginal |
Vaginal remnant will be presentinambiguous genitalia |
|
|
|
8 |
Masculinisation externally complete by12 weeks but growing penis needs androgen up to 40 weeks |
Absent androgens after this (in |
|
|
|
9 |
“hypospadias” assumes male gender, therefore only use if scrotum fusedand testes descended |
‘hypospadias’ = boy with normal hormones and local anomaly of urethral development. If UDT/bifid scrotum full DSD workup is required |
|
|
|
10 |
Nonhormonal genital anomalies outsidenarrow spectrum from male to female |
Perineal anatomical anomalies includegenitalia as well as other features (e.g. abdominal wall or anorectal defects) |
References
1. Hutson, J.: Development of the Urogenital System. In: Gray's Anatomy (4th edn). Edited by S. Standring.London, vol. Chapter 78, pp. 1305-1325, 2008
2. Stephens, F., Smith, E., Hutson, J.M..: Congenital Anomalies of the Kidney, Urinary and Genital Tracts, 2nd Edn, Martin Dunitz, London, 2002
3. Warne, G. L., Kanumakala, S.: Molecular endocrinology of sex differentiation. Semin Reprod Med, 20: 169, 2002
4. Tong, S. Y., Hutson, J. M., Watts, L. M.: Does testosterone diffuse down the wolffian duct during sexual differentiation? J Urol, 155: 2057, 1996
5. Grumbach, M., Hughes, I., Conte, F.: Disorders of Sex Differentiation. In: Williams Textbook of Endocrinology, 10th ed. Edited by K. H. Larsen PR, Melmed S, Polonsky KS. Philadelphia: WB Saunders, pp. 842-968, 2003
6. Nef, S., Parada, L. F.: Cryptorchidism in mice mutant for Insl3. Nat Genet, 22: 295, 1999
7. Zimmerman, S., Steding, G., Emmen, J. et al.: Targeted disruption of the INSL3 gene causes bilateral cryprotchidism. Mol. Endocrinol., 13: 681, 1999
8. Adham, I. M., Agoulnik, A. I.: Insulin-like 3 signalling in testicular descent. Int J Androl, 27: 257, 2004
9. Hutson, J. M., Hasthorpe, S.: Testicular descent and cryptorchidism: the state of the art in 2004. J Pediatr Surg, 40: 297, 2005
10. Penington, E. C., Hutson, J. M.: The urethral plate--does it grow into the genital tubercle or within it? BJU Int, 89: 733, 2002
11. Miller, A., Hong, M. K., Hutson, J.: The broad ligament: a review of its anatomy and development in different species and hormonal environments. Clinical Anatomy, 17: 244, 2004
12. Clarnette, T., Sugita, Y., Hutson, J.M..: Genital anomalies in the human and animal models reveal the mechanisms and hormones governing testicular descent. Brit J Urol, 79: 99, 1997
13. Low, Y., Hutson, J. M.: Rules for clinical diagnosis in babies with ambiguous genitalia. J Paediatr Child Health, 39: 406, 2003
14. Kaplan, S. L., Grumbach, M. M., Aubert, M. L.: The ontogenesis of pituitary hormones and hypothalamic factors in the human fetus: maturation of central nervous system regulation of anterior pituitary function. Recent Prog Horm Res, 32: 161, 1976
15. Hutson, J.M, Chow, C.W, Ng, W.: Persistent mullerian duct syndrome with transverse testicular extopia. anexperiment of nature with clues for understanding testicular descent. Pediatr Surg Int, 2: 191, 1987
16. Hutson, J. M., Davidson, P., Reece, L. et al.: Failure of gubernacular development in the persistent mullerian duct syndrome allows herniation of the testes. Pediatr Surg Int, 9: 544, 1994
17. Hutson, J. M., Baker, M.: A hypothesis to explain abnormal gonadal descent in persistent mullerianduct syndrome. Pediatric Surgery International, 9: 542, 1994
18. Josso, N., Fekete, C., Cachin, O. et al.: Persistence of Mullerian ducts in male pseudohermaphroditism, and its relationship to cryptorchidism. Clin Endocrinol (Oxf), 19: 247, 1983
19. Griffin, J. E., Wilson, J. D.: Syndromes of androgen resistance. Hosp Pract (Off Ed), 22: 159, 1987
20. Hutson, J. M.: A biphasic model for the hormonal control of testicular descent. Lancet, 2: 419, 1985
21. Hutson, J. M.: Testicular feminization: a model for testicular descent in mice and men. J Pediatr Surg, 21: 195, 1986
22. Vidal, I., Gorduza, D. B., Haraux, E. et al.: Surgical options in disorders of sex development (dsd) with ambiguous genitalia. Best Pract Res Clin Endocrinol Metab, 24: 311
23. Houk, C. P., Hughes, I. A., Ahmed, S. F. et al.: Summary of consensus statement on intersex disorders and their management. International Intersex Consensus Conference.Pediatrics, 118: 753, 2006
24. Lean, W. L., Deshpande, A., Hutson, J.M. et al.: Cosmetic and anatomic outcomes after feminizing surgery for ambiguous genitalia. J Pediatr Surg, 40: 1856, 2005
25. Minto, C. L., Liao, L. M., Woodhouse, C. R. et al.: The effect of clitoral surgery on sexual outcome in individuals who have intersex conditions with ambiguous genitalia: a cross-sectional study. Lancet, 361: 1252, 2003
26. Rangecroft, L.: Surgical management of ambiguous genitalia. Arch Dis Child, 88: 799, 2003
27. Creighton, S. M., Minto, C. L., Steele, S. J.: Objective cosmetic and anatomical outcomes at adolescence of feminising surgery for ambiguous genitalia done in childhood. Lancet, 358: 124, 2001
28. Warne, G., Grover, S., Hutson, J.M. et al.: A long-term outcome study of intersex conditions. J Pediatr Endocrinol Metab, 18: 555, 2005
29. Crawford, J. M., Warne, G., Grover, S. et al.: Results from a pediatric surgical centre justify early intervention in disorders of sex development. J Pediatr Surg, 44: 413, 2009
30. Hrabovszky, Z., Hutson, J. M.: Surgical treatment of intersex abnormalities: a review. Surgery, 131: 92, 2002
31. Hendren, W. H.: Surgical approach to intersex problems. Semin Pediatr Surg, 7: 8, 1998
32. Kogan, S. J.: Feminizing genital reconstruction for male pseudohermaphroditism. Eur J Pediatr, 152 Suppl 2: S85, 1993
33. Gonzalez, R., Fernandes, E. T.: Single-stage feminization genitoplasty. J Urol, 143: 776, 1990
34. Duckett, J. W., Baskin, L. S.: Genitoplasty for intersex anomalies. Eur J Pediatr, 152 Suppl 2: S80, 1993
35. Hutson, J.M., Voigt, R., Luthra, M. et al.: Girth-reduction clitoroplasty - a new technique: experience with 37 patients. Pediatric Surgery International, 6: 336, 1991
36. Roberts, J. P., Hutson, J. M.: Reduction of scrotalized skin improves the cosmetic appearance of feminising genitoplasty. Pediatr Surg Int, 12: 228, 1997
37. Donahoe, P. K., Hendren, W. H., 3rd: Perineal reconstruction in ambiguous genitalia infants raised as females. Ann Surg, 200: 363, 1984
38. Passerini-Glazel, G.: A new 1-stage procedure for clitorovaginoplasty in severely masculinized female pseudohermaphrodites. J Urol, 142: 565, 1989.
39. Hutson, J.M., Warne, G.L., Grover, S.R. Disorders of Sex Development: An integrated approach to management. Springer, Berlin, 2012.
40. Hutson, J.M., Grover, S.R., O’Connell, M., Pennell, S.D. Malformation syndromes associated with disorders of sex development. Nat Rev Endocrinol., 10(8)476-87 (2014).
Figure Legend
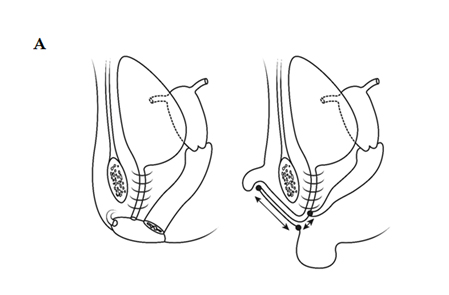
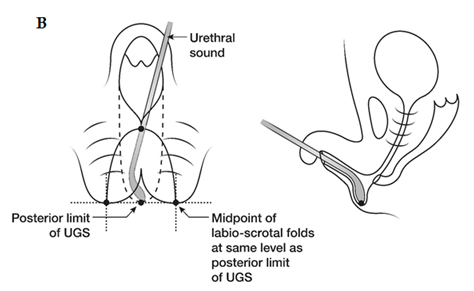
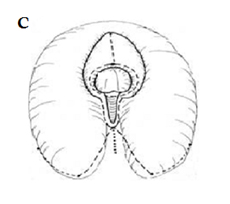
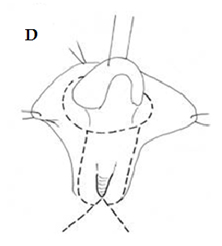
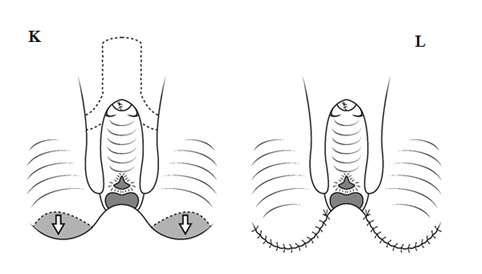
Fig. A. Relative anatomy of normal female versus virilised female in CAH, with masculinisation of anterior urethra pushing opening on perineum forward, even though join between urogenital sinus and vagina remains quite close to surface. B. Marking the skin flaps for (V-Y) vaginoplasty using a sound to mark the posterior limit of the masulinised urogenital sinus. This marks the base of the skin flap so that it aligns with the vagina and the posterior limit of the labio-scrotal folds. C. Skin incisions for clitoroplasty, vaginoplasty, and redundant labia. D. Stay sutures in glans and prepuce. Incision lines on ventral surface are shown in detail in relation to the ventral mucosal strip of the urogenital sinus and to vaginoplasty (Y-V). E. Mobilisation of phallic shaft ventrally to free the urogenital mucosal strip (X) and protect the underlying urethra where it passes between the crura (Y). Dorsal mobilisation of shaft skin to expose and divide the suspensory ligament (Z). F. Caudal traction on phallus to expose bifurcation of crura and periosteum of symphysis pubis (W). A right-angled divulser is pushed through the bifurcation, with care to avoid the urethra, to apply rubber catheter tourniquets to each crus. G. Tourniquets around each crus, showing mucosal strip and urethra retracted to safety. H. Anterior portion of shaft and glans is excised with tourniquet control, avoiding the dorsal nerves and vessels. Extra bulk can be removed by “filleting” out erectile tissue from the cut surface. I. Suture of phallus into hair-pin bend by approximating raw surfaces (excluding distal frenular area) and suturing apposed lateral cut edges of tunica. J. Folded phallus sutured to pubic periosteum. The anterior midline fold of the clitoral skin is sutured to the shaft. The skin edge (A1) is sutured to the flange of subcoronal skin (A2). Preputial flaps are sutured along each side of the re-attached vestibular mucosal strip to simulate labia minora (B1-B2, C1-C2). K. The removal of the posterior end of the labioscrotal folds. Final result at end of operation. The Y-V vaginoplasty flap and excision of redundant labial skin are also shown.
 |
 |
 |
 |
 |
 |
 |
 |
